Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cardiac & Cardiovascular Systems
Cardiac surgeries have been improved by accompanying developing cardioplegia solutions. However, the cardioplegia application presents an ongoing challenge with a view of a sufficiently restored cardiac function.
- cardioplegia
- IR injury
- ion transport
- ion channel
- cardiochannelopathy
1. Role of Cardioplegia and Potential Injuries
During cardiac surgery, hyperkalemic cardioplegic solutions are the essential gold standard to prevent cardiac depolarization and temporarily sustain a cardiac arrest through the reduction of cardiac loading. In detail, elevated extracellular K+ concentrations (10–40 mM) of hyperkalemic cardioplegic solutions increase the resting myocyte membrane potentials (Em) from −85 mV to between −65 and −40 mV, which inactivates fast Na+ channels [1]. These higher potentials block the conduction of myocardial action potentials, induce a depolarized arrest, and minimize tissue damage during cardiac surgery. Various cardioplegic solutions based on the component, temperature, or period of delivery have been developed as protective strategies.
Although cardioplegic solutions to protect cardiac cell death from ischemia, detrimental injury may occur during intraoperative ischemia due to multi-dose infusions of a cardioplegic solution or the misdistribution of a solution distal to total coronary occlusions [2]. In addition, there is the potential for a reperfusion injury during each infusion and after the aortic cross-clamp removal [2]. In the case of del Nido cardioplegia, the intracellular pH is maintained and the influx of Ca2+ during ischemic arrest is reduced; this has been used to improve myocardial protection [3,4]. However, although del Nido cardioplegia does not lead to clinical side effects, there is an adverse effect. When del Nido cardioplegia was used in an adult aortic root surgery, the ischemic time of the patient was increased compared with blood-based cardioplegia, thereby increasing the myocardial injury Although cardioplegic solutions possess advantages such as successful cardiac surgery to protect an IR injury, diabetic hearts have been shown to have worse clinical outcomes compared with non-diabetic hearts [5,6,7].
2. Cardioplegia Solution Characteristics, Risk Factors, and Related Mechanisms
2.1. Myocardial Ischemia/Reperfusion Injuries
Myocardial ischemia/reperfusion (IR) injuries occur during cardiac surgery and cardioplegic solutions have a protective effect although their effects are limited [8]. In the ischemic state, a lack of oxygen in cells results in ATP and lactic acid production by anaerobic glycolysis and, thus, a reduction in the intracellular pH [9]. Furthermore, the lactic acid generated inhibits the functions of mitochondrial permeability transition pores (mPTPs, a non-selective channel in the mitochondria) and myofibril contracture [10,11]. During a myocardial reperfusion, Ca2+ levels increase in the cardiomyocytes due to ROS-induced sarcoplasmic reticulum (SR) dysfunction [11,12], resulting in a reperfusion injury. The pH recovery mediates the opening of mPTPs, which interferes with mitochondrial membrane potentials [10]. Reoxygenation by reperfusion also increases the cytosolic Ca2+ levels in cardiomyocytes, causing their hypercontraction and subsequent cell death [12,13,14]. A schematic illustration of the mechanism of an IR injury is represented in Figure 1.
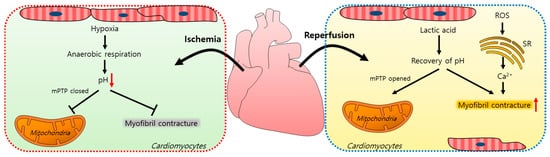
Figure 1. Schematic illustration of the mechanism of an ischemia/reperfusion injury in cardiomyocytes. Ischemia-induced hypoxia results in anaerobic respiration and reduces the intracellular pH, which closes mPTPs and reduces myofibril contracture. During reperfusion, lactic acid accumulates, the intracellular pH increases, and mPTPs open. ROS increase the intracellular Ca2+ release from the SR and induce myofibril contracture. mPTP: mitochondrial permeability transition pore; ROS: reactive oxygen species; SR: sarcoplasmic reticulum.
2.2. Cardioplegia-Mediated Apoptosis
In this section, we discuss the mechanism of cardioplegia application and associated injuries after a cardioplegic arrest and summarize the numerous studies undertaken to improve the cardioprotective methods against an IR injury over the past several decades. Cold crystalloid or blood cardioplegic solutions have been widely used for cardiac surgery for myocardial protection. However, it has been addressed that cold crystalloid cardioplegia induces early apoptosis signaling events in the myocardial endothelium and cardiomyocytes [15,16]. Two types of cardioplegia methods have been evaluated after a cardioplegic arrest on myocardial injuries in dogs. Both cold blood and cold crystalloid cardioplegia resulted in higher cardiomyocyte apoptosis percentages and an increased caspase-3 expression [17]. To reduce the threat points posed by a cardioplegic arrest, the NHE-1 inhibitor eniporide was applied in a cold cardioplegic or a Krebs–Ringer solution to guinea pigs and was found to improve the relationship of the diastolic left ventricular pressure intracellular Ca2+ levels [18]. The cardioplegia group treated with eniporide showed reduced Ca2+ loading and infarct sizes [18]. Furthermore, the application of cyclosporine A, a mitochondrial permeability transition pore inhibitor, in the presence of cold crystalloid cardioplegia prevented mitochondrial permeability, the mitochondrial translocation of Bax, and protected the mitochondrial structures in a newborn piglet myocardium [16]. Bax (a member of the Bcl-2 family) triggers the permeabilization of the mitochondria to form pores on the mitochondrial outer membrane; this was increased by cold crystalloid cardioplegia. This translocation of Bax was inhibited by a pretreatment with cyclosporine A. The role of AMP-activated protein kinase (AMPK) on endoplasmic reticulum (ER) stress has been evaluated in a cardioplegic-induced hypoxia/reoxygenation (H/R) injury [19]. AMPK activation dramatically attenuated the H/R-induced apoptosis of cardiomyocytes by increasing the anti-apoptotic (e.g., Grp 78 protein) levels and suppressing the pro-apoptotic signals (e.g., caspase-12 and GADD153 (growth arrest and DNA damage-inducible gene 153)) [19]. The administration of bradykinin (BK) during cold crystalloid cardioplegia decreased oxygen consumption and reduced the myocardial metabolic demands of mild-to-moderate hypothermia [20,21] as well as IR-induced inflammation and apoptosis whereas the inhibition of the BK receptor or nitric oxide synthase diminished the inflammatory and apoptotic responses of rabbit cardiomyocytes [22]. Furthermore, nitric oxide (NO) modulation during a cardioplegic arrest might improve the anti-apoptotic signal profile through the NF-κB/Akt/Bcl-2/Bax axis [19]. More recently, the potential protective effects of the flavonoids astragalin and dihydromyricetin in ischemia during a cold crystalloid cardioplegic arrest were evaluated [23]. A cold crystalloid cardioplegic solution in the presence of astragalin or dihydromyricetin enhanced the anti-inflammatory pathways and attenuated the oxidative signals [23]. In addition to mitochondrial stress, the expression of urocortin (a cardioprotective protein) was investigated under a warm blood cardioplegic arrest [24,25]. More recently, in the heart of a diabetic patient, the downregulation of urocortin was associated with cardiomyocyte apoptosis and the alteration of the nuclear location of PKC-δ [26,27]. Cardioplegia techniques were also compared with the aim of reducing the risk of an IR injury. Retrograde cardioplegia involves the cannulation of the solution into the coronary sinus whereas antegrade cardioplegia involves cannulation into the aortic root [28]. Retrograde cardioplegia causes greater cardiomyocyte apoptosis and caspase-3 activation than antegrade cardioplegia after IR [29]. Although changes in the strategies of cardioplegic solutions have been addressed, there are no clear advantages of one cardioplegic strategy over another. Accordingly, we propose an understanding of the cardioplegia-occurring molecular mechanism with a view of a cellular pathological response.
2.3. Cardioplegia-Induced Inflammation and Dysfunction
Cytokine levels play critical roles in inflammatory responses. It is well-known that a cardiopulmonary bypass (CPB) contributes to postoperative complications [30]. The levels of tumor necrosis factor (TNF)-α, interleukin (IL)-6/8, and IL-10 in the blood of patients that had received cold crystalloid or warm blood cardioplegia were compared [30]. They reported that the serum levels of TNF-α, IL-6, and IL-8—excluding IL-10—were higher in the cold crystalloid than in the warm blood-exposed group. Based on their comparative study of cardioplegia trials, it would seem that warm blood cardioplegia may reduce the inflammatory response more effectively than cold crystalloid cardioplegia. Furthermore, an additional supplementation of an amino acid such as L-arginine in cardioplegia reduced the cytokine release; this was significant for IL-6 [31]. In addition to the inflammatory response, hyperkalemic cardioplegia induced cardiomyocyte swelling and reduced contractility in various models [32,33,34]. Treatment with the adenosine triphosphate-sensitive potassium channel (KATP) opener diazoxide also reduced cardiomyocyte swelling and inhibited cardioplegia-mediated reduced contractility [33]. It was revealed that the maintenance of the cardiomyocyte volume was associated with improved contractility and stunning.
2.4. Cardioplegia-Induced Oxidative Stress
Oxidative stress induced by CPB mediates myocardium inflammation and damage. To reduce the effect of oxidative stress, a pyruvate-enriched cardioplegia was investigated in pigs [35]. A combination of blood and cold crystalloid cardioplegia with pyruvate increased the tissue inhibitor of metalloproteinase-2 (TIMP-2) contents (a known stimulator of cell migration and apoptosis) and inhibited the metalloproteinase-9 (MMP-9) activity [35]. In addition, the application of pyruvate during cardioplegia reduced CPB-induced myocardial inflammation by increasing the myocardial GSH redox state (GSH/GSSG) and TIMP-2 levels [35]. GSH and GSSG are the mediators of oxidation/reduction reactions and the GSH/GSSG ratio is used as a marker of oxidative stress [36]. Cardioplegia with pyruvate decreased this ratio in the coronary sinus and finally reduced oxidative stress [35]. Moreover, a comparison of inflammatory cytokines after the administration of two types of cardioplegia solution, i.e., a “del Nido” or a “modified St. Thomas” solution, revealed no detectable difference, suggesting that the consideration of the administration intervals is more important than the cardioplegia solution type [37].
Various studies have been performed to improve the arrested status and to abolish the risk factors of cardioplegia such as inflammation, swelling, stunning, and oxidative stress (summarized in Table 1). However, a more effective strategy is still required to properly address these complications. The precise study of the mechanisms responsible for the effects of administered compounds on cardioplegia is required. In particular, an understanding of the changes in electrolytes through ion channels/transporters and the modulations of water channels would aid the understanding of heart damage. In the following section, we focus on the basic concept of electrolyte homeostasis as well as the changes of channel activity in cardiac tissue.
Table 1. Characteristics of the modified cardioplegia solutions.
| Drugs | Solutions | Limits | Mechanisms | Ref. |
|---|---|---|---|---|
| Adenosine triphosphate-sensitive K+ channel (KATP) opener diazoxide | Hyperkalemic cardioplegia | Myocyte swelling and reduced contractility | Diazoxide prevented myocyte swelling and reduced contractility by blocking the KATP channel related to myocardial stunning | [33] |
| Cyclosporine A (MPTP-opening inhibitor) |
Cold antegrade crystalloid cardioplegia | Apoptosis-related alteration in the mitochondrial structure | Promoted Bax translocation and inhibited calcineurin related to Ca2+ homeostasis | [16] |
| AMPK activator (AICAR, metformin) | Deoxygenated hypothermic cardioplegia | Cardiomyocytic apoptosis |
Enhanced anti-apoptotic proteins (Grp78) and decreased ER stress | [19] |
| Pyruvate-enriched cardioplegia |
Crystalloid cardioplegia | Inflammation that can damage the myocardium | Attenuated oxidative stress during CPB and increased TIMP-2 | [35] |
| L-arginine cardioplegia | Cold blood + anterograde and retrograde cardioplegia | Myocardial ischemic damage | Production of nitric oxide, increased IL-2 receptor, IL-6, and tumor necrosis factor levels | [31] |
| Bradykinin | Cold crystalloid cardioplegia | Apoptosis under cardiopulmonary bypass | Decreased NO level and nuclear translocation of NF-κB | [19] |
| Dilong (earthworm) | High KCl cardiologic solution | Apoptosis of cardiomyoblast (H9c2 cells) | Attenuated caspase-3 activation and enhanced PI3K/Akt and Bcl-2 | [38] |
| Urocortin | Cold blood cardioplegia | Apoptosis and dysfunction in diabetic hearts | Induction and mitochondrial relocation of PKC-δ | [27] |
| NHE-1 inhibitor eniporide |
Cold cardioplegia | Ca2+ overloading | Inhibition of NHE-1 reduced the infarct size after hypothermic ischemia | [18] |
KATP: adenosine triphosphate-sensitive potassium channel; MPTP: mitochondrial permeability transition pore; Bax: Bcl-2-associated X; AMPK: AMP-activated protein kinase; AICAR: 5-aminoimidazole-4-carboxamide ribonucleoside; Grp78: glucose-regulated protein; ER: endoplasmic reticulum; CPB: cardiopulmonary bypass; TIMP-2: tissue inhibitor of metalloproteinase 2; IL-2 receptor: interleukin-2 receptor; IL-6: interleukin-6; NO: nitric oxide; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; PI3K/Akt: phosphoinositide-3-kinase/protein kinase B; PKC-δ: protein kinase C-δ; NHE-1: Na+-H+ exchanger-1.
This entry is adapted from the peer-reviewed paper 10.3390/antiox10121878
This entry is offline, you can click here to edit this entry!