Cisplatin, also referred to as cis-diamminedichloroplatinum (II) (CDDP), is a widely used, Pt-based anti-cancer chemotherapeutic agent. This transition metal coordination complex has a square planar molecular geometry and presents as a solid, yellow powder at room temperature. CDDP is fairly insoluble in most substances; however, it can be dissolved in dimethylformamide, and is somewhat soluble in water, but most preferable in saline sodium. In solid form, CDDP is stable for about two years if stored at room temperature in dry conditions and light protected; however, it is known to convert slowly to its trans-form. The stability of CDDP in sodium chloride solution is dependent on the chloride ion concentration; it is more stable when suspended in normal saline (0.9% sodium chloride). Saline also provides a reno-protective function, increasing hydration and aiding in excretion of CDDP from the kidneys.
- cisplatin
- carboplatin
- oxaliplatin
- cellular uptake
1. Clinical Usage of Cisplatin
| Platinating Agent | Indication | Major Side Effect |
|---|---|---|
| Cisplatin | Breast, ovarian, testicular, head-and-neck, esophageal, lung, bladder, brain tumours | Nephrotoxicity, ototoxicity, neurotoxicity, hepatotoxicity, cardiotoxicity |
| Carboplatin | Ovarian, lung, certain head-and-neck | Myelosuppression (thrombocytopenia) |
| Oxaliplatin | Colorectal | Peripheral neurotoxicity, Hepatic Sinusoidal obstruction syndrome, myelosuppression |
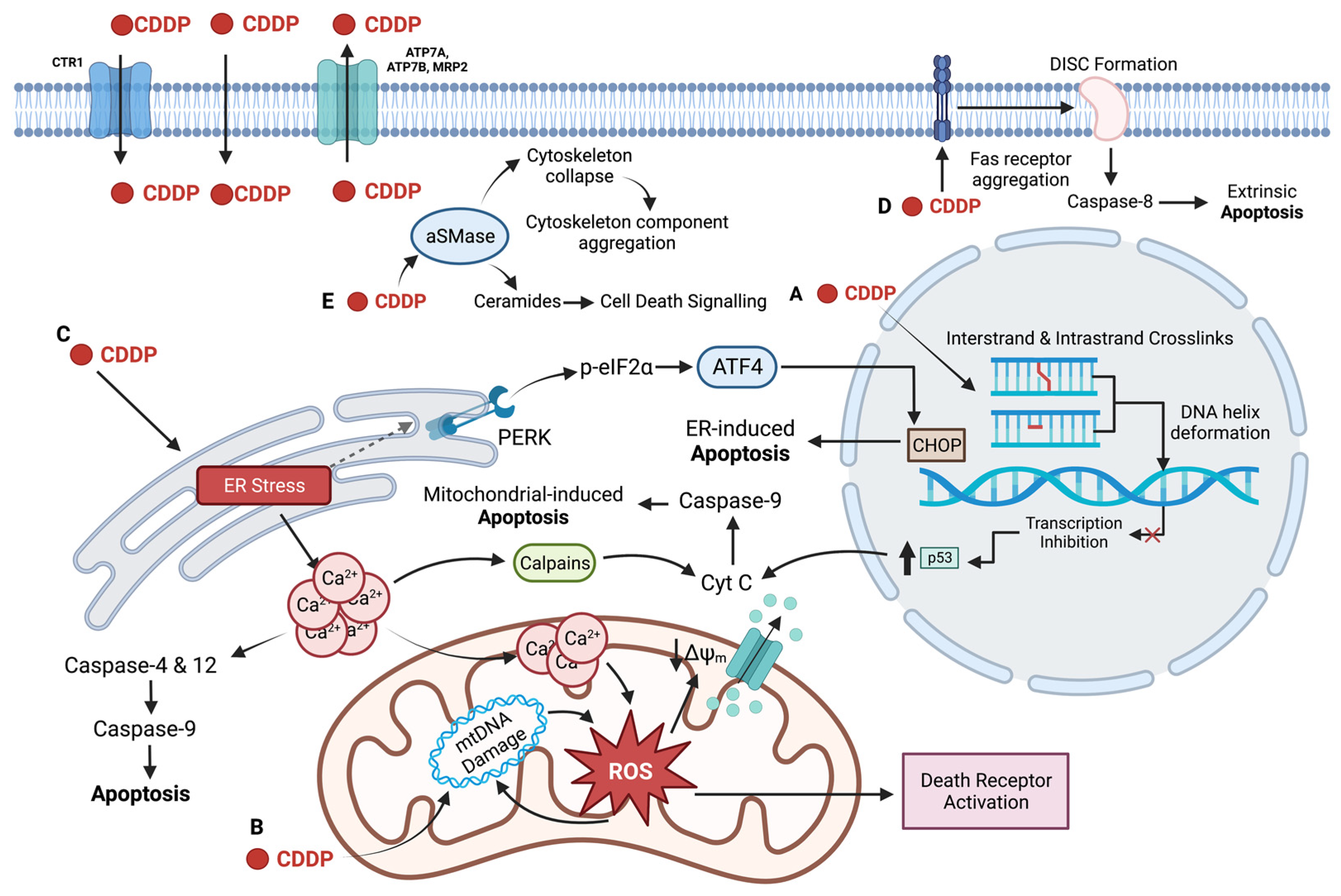
2. Cellular Uptake of Cisplatin
3. DNA as the Primary Target of Cisplatin
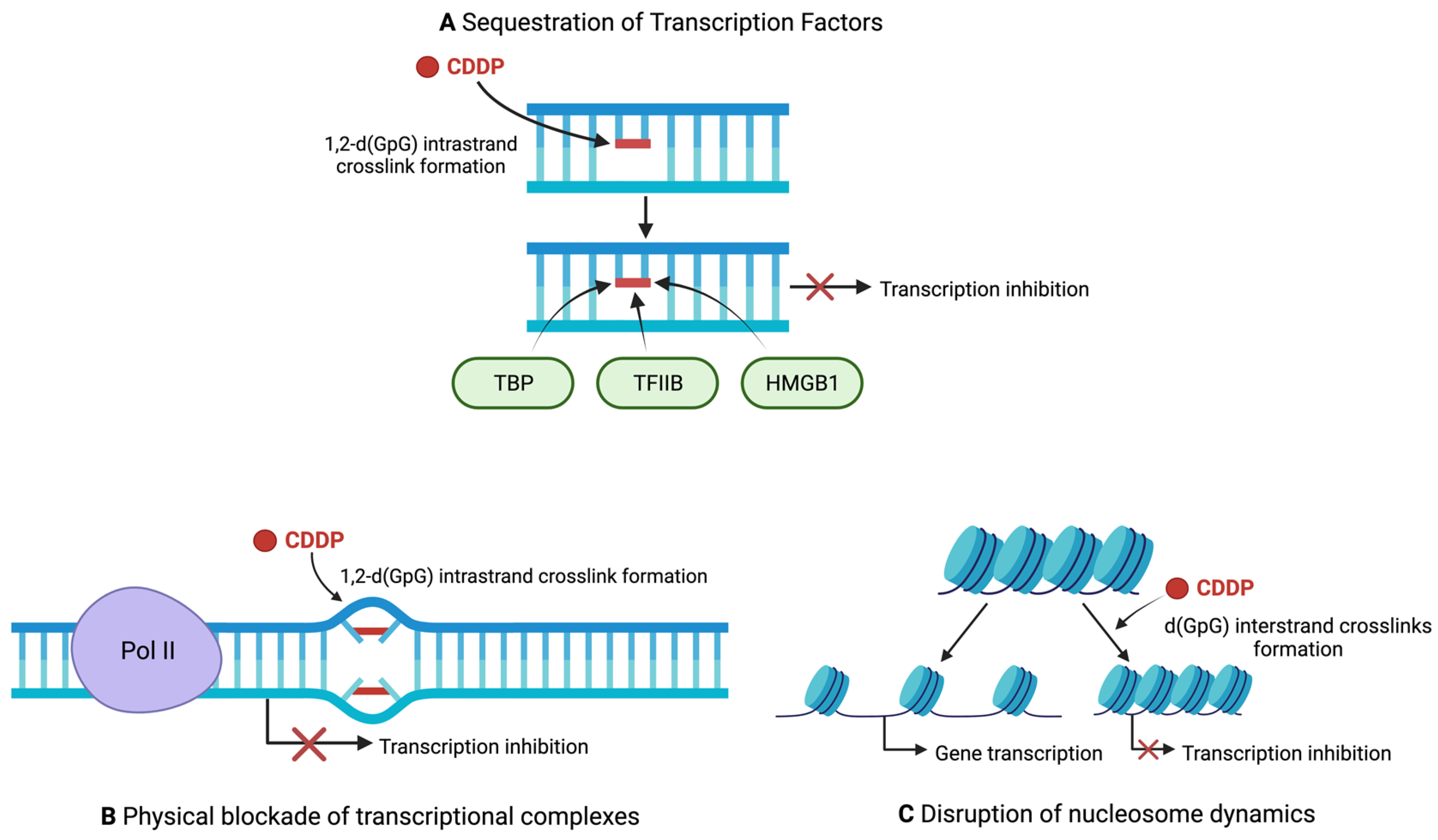
4. Regulation of Transcription by Cisplatin
5. Non-Nuclear Targets of Cisplatin
6. Limitations of the Use of Cisplatin
This entry is adapted from the peer-reviewed paper 10.3390/ijms232315410
References
- Jamieson, E.R.; Lippard, S.J. Structure, Recognition, and Processing of Cisplatin−DNA Adducts. Chem. Rev. 1999, 99, 2467–2498.
- Einhorn, L.H. Treatment of testicular cancer: A new and improved model. J. Clin. Oncol. 1990, 8, 1777–1781.
- Fung, C.; Dinh, P.C.; Fossa, S.D.; Travis, L.B. Testicular Cancer Survivorship. J. Natl. Compr. Cancer Netw. 2019, 17, 1557–1568.
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378.
- Boulikas, T.; Vougiouka, M. Recent clinical trials using cisplatin, carboplatin and their combination chemotherapy drugs (review). Oncol. Rep. 2004, 11, 559–595.
- DeConti, R.C.; Toftness, B.R.; Lange, R.C.; A Creasey, W. Clinical and pharmacological studies with cis-diamminedichloroplatinum (II). Cancer Res. 1973, 33, 1310–1315.
- Gale, G.R.; Morris, C.R.; Atkins, L.M.; Smith, A.B. Binding of an antitumor platinum compound to cells as influenced by physical factors and pharmacologically active agents. Cancer Res. 1973, 33, 813–818.
- Eljack, N.D.; Ma, H.-Y.M.; Drucker, J.; Shen, C.; Hambley, T.W.; New, E.J.; Friedrich, T.; Clarke, R.J. Mechanisms of cell uptake and toxicity of the anticancer drug cisplatin. Metallomics 2014, 6, 2126–2133.
- Binks, S.P.; Dobrota, M. Kinetics and mechanism of uptake of platinum-based pharmaceuticals by the rat small intestine. Biochem. Pharmacol. 1990, 40, 1329–1336.
- Andrews, P.A.; Mann, S.C.; Velury, S.; Howell, S.B. Cisplatin Uptake Mediated Cisplatin-Resistance in Human Ovarian Carcinoma Cells. In Platinum and Other Metal Coordination Compounds in Cancer Chemotherapy: Proceedings of the Fifth International Symposium on Platinum and Other Metal Coordination Compounds in Cancer Chemotherapy Abano, Padua, Italy, 29 June–2 July 1987; Nicolini, M., Ed.; Springer: Boston, MA, USA, 1988; pp. 248–254.
- Andrews, P.A.; Albright, K.D. Role of Membrane Ion Transport in Cisplatin Accumulation. In Platinum and Other Metal Coordination Compounds in Cancer Chemotherapy; Howell, S.B., Ed.; Springer: Boston, MA, USA, 1991; pp. 151–159.
- Ivy, K.D.; Kaplan, J.H. A Re-Evaluation of the Role of hCTR1, the Human High-Affinity Copper Transporter, in Platinum-Drug Entry into Human Cells. Mol. Pharmacol. 2013, 83, 1237–1246.
- Mann, S.C.; Andrews, P.A.; Howell, S.B. Short-term cis-diamminedichloroplatinum(II) accumulation in sensitive and resistant human ovarian carcinoma cells. Cancer Chemother Pharmacol. 1990, 25, 236–240.
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302.
- Katano, K.; Kondo, A.; Safaei, R.; Holzer, A.; Samimi, G.; Mishima, M.; Kuo, Y.-M.; Rochdi, M.; Howell, S.B. Acquisition of resistance to cisplatin is accompanied by changes in the cellular pharmacology of copper. Cancer Res. 2002, 62, 6559–6565.
- Holzer, A.K.; Samimi, G.; Katano, K.; Naerdemann, W.; Lin, X.; Safaei, R.; Howell, S.B. The Copper Influx Transporter Human Copper Transport Protein 1 Regulates the Uptake of Cisplatin in Human Ovarian Carcinoma Cells. Mol. Pharmacol. 2004, 66, 817–823.
- Holzer, A.K.; Katano, K.; Klomp, L.W.J.; Howell, S.B. Cisplatin Rapidly Down-regulates Its Own Influx Transporter hCTR1 in Cultured Human Ovarian Carcinoma Cells. Clin. Cancer Res. 2004, 10, 6744–6749.
- Petris, M.J.; Smith, K.; Lee, J.; Thiele, D.J. Copper-stimulated Endocytosis and Degradation of the Human Copper Transporter, hCtr1. J. Biol. Chem. 2003, 278, 9639–9646.
- Kalayda, G.V.; Wagner, C.H.; Jaehde, U. Relevance of copper transporter 1 for cisplatin resistance in human ovarian carcinoma cells. J. Inorg. Biochem. 2012, 116, 1–10.
- Sinani, D.; Adle, D.J.; Kim, H.; Lee, J. Distinct Mechanisms for Ctr1-mediated Copper and Cisplatin Transport. J. Biol. Chem. 2007, 282, 26775–26785.
- Beretta, G.L.; Gatti, L.; Tinelli, S.; Corna, E.; Colangelo, D.; Zunino, F.; Perego, P. Cellular pharmacology of cisplatin in relation to the expression of human copper transporter CTR1 in different pairs of cisplatin-sensitive and -resistant cells. Biochem. Pharmacol. 2004, 68, 283–291.
- Hall, M.D.; Okabe, M.; Shen, D.-W.; Liang, X.-J.; Gottesman, M.M. The Role of Cellular Accumulation in Determining Sensitivity to Platinum-Based Chemotherapy. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 495–535.
- Song, I.S.; Savaraj, N.; Siddik, Z.H.; Liu, P.; Wei, Y.; Wu, C.J.; Kuo, M.T. Role of human copper transporter Ctr1 in the transport of platinum-based antitumor agents in cisplatin-sensitive and cisplatin-resistant cells. Mol. Cancer Ther. 2004, 3, 1543–1549.
- A Andrews, P.; Mann, S.C.; Huynh, H.H.; Albright, K.D. Role of the Na+, K(+)-adenosine triphosphatase in the accumulation of cis-diamminedichloroplatinum(II) in human ovarian carcinoma cells. Cancer Res. 1991, 51, 3677–3681.
- Kishimoto, S.; Kawazoe, Y.; Ikeno, M.; Saitoh, M.; Nakano, Y.; Nishi, Y.; Fukushima, S.; Takeuchi, Y. Role of Na+, K+-ATPase α1 subunit in the intracellular accumulation of cisplatin. Cancer Chemother. Pharmacol. 2006, 57, 84–90.
- A Andrews, P.; Albright, K.D. Mitochondrial defects in cis-diamminedichloroplatinum(II)-resistant human ovarian carcinoma cells. Cancer Res. 1992, 52, 1895–1901.
- Walker, W.F.; Johnston, I.D.A. 2—Water and Electrolyte Metabolism. In The Metabolic Basis of Surgical Care; Walker, W.F., Johnston, I.D.A., Eds.; Butterworth-Heinemann: Oxford, UK, 1971.
- El-Khateeb, M.; Appleton, T.G.; Gahan, L.R.; Charles, B.G.; Berners-Price, S.J.; Bolton, A.-M. Reactions of cisplatin hydrolytes with methionine, cysteine, and plasma ultrafiltrate studied by a combination of HPLC and NMR techniques. J. Inorg. Biochem. 1999, 77, 13–21.
- Pinto, A.L.; Lippard, S.J. Binding of the antitumor drug cis-diamminedichloroplatinum(II) (cisplatin) to DNA. Biochim. Biophys. Acta 1985, 780, 167–180.
- Miller, S.E.; House, D.A. The hydrolysis products of cis-diamminedichloroplatinum(II) 5. The anation kinetics of cis-Pt(X)(NH3)2(OH2)+ (XCl, OH) with glycine, monohydrogen malonate and chloride. Inorganica Chim. Acta 1991, 187, 125–132.
- Kelland, L.R. New platinum antitumor complexes. Critical Rev. Oncol. /Hematol. 1993, 15, 191–219.
- Eastman, A. Characterization of the adducts produced in DNA by cis-diamminedichloroplatinum(II) and cis-dichloro(ethylenediamine)platinum(II). Biochemistry 1983, 22, 3927–3933.
- Plooy, A.C.; Fichtinger-Schepman, A.M.J.; Schutte, H.H.; Van Dijk, M.; Lohman, P.H. The quantitative detection of various Pt-DNA-adducts in Chinese hamster ovary cells treated with cisplatin: Application of immunochemical techniques. Carcinogenesis 1985, 6, 561–566.
- Eastman, A. The formation, isolation and characterization of DNA adducts produced by anticancer platinum complexes. Pharmacol. Ther. 1987, 34, 155–166.
- Giraud-Panis, M.-J.; Malinge, J.-M.; Sedletska, Y. Cisplatin Is a DNA-Damaging Antitumour Compound Triggering Multifactorial Biochemical Responses in Cancer Cells: Importance of Apoptotic Pathways. Curr. Med. Chem. Agents 2005, 5, 251–265.
- Beck, D.J.; Brubaker, R.R. Effect of cis-platinum(II)diamminodichloride on wild type and deoxyribonucleic acid repair deficient mutants of Escherichia coli. J. Bacteriol. 1973, 116, 1247–1252.
- Fraval, H.; Rawlings, C.; Roberts, J. Increased sensitivity of UV-repair-deficient human cells to DNA bound platinum products which unlike thymine dimers are not recognized by an endonuclease extracted from Micrococcus luteus. Mutat. Res. Mol. Mech. Mutagen. 1978, 51, 121–132.
- Harder, H.C.; Smith, R.G.; Leroy, A.F. Template primer inactivation by cis- and trans-dichlorodiammine platinum for human DNA polymerase alpha, beta, and Rauscher murine leukemia virus reverse transcriptase, as a mechanism of cytotoxicity. Cancer Res. 1976, 36, 3821–3829.
- Pinto, A.L.; Lippard, S.J. Sequence-dependent termination of in vitro DNA synthesis by cis- and trans-diamminedichloroplatinum (II). Proc. Natl. Acad. Sci. USA 1985, 82, 4616–4619.
- Sorenson, C.M.; Eastman, A. Mechanism of cis-diamminedichloroplatinum(II)-induced cytotoxicity: Role of G2 arrest and DNA double-strand breaks. Cancer Res. 1988, 48, 4484–4488.
- Sorenson, C.M.; Eastman, A. Influence of cis-diamminedichloroplatinum(II) on DNA synthesis and cell cycle progression in excision repair proficient and deficient Chinese hamster ovary cells. Cancer Res. 1988, 48, 6703–6707.
- Sorenson, C.M.; Barry, M.A.; Eastman, A. Analysis of Events Associated With Cell Cycle Arrest at G2 Phase and Cell Death Induced by Cisplatin. Gynecol. Oncol. 1990, 82, 749–755.
- Jung, Y.; Lippard, S.J. Multiple States of Stalled T7 RNA Polymerase at DNA Lesions Generated by Platinum Anticancer Agents. J. Biol. Chem. 2003, 278, 52084–52092.
- Tornaletti, S.; Patrick, S.M.; Turchi, J.J.; Hanawalt, P.C. Behavior of T7 RNA Polymerase and Mammalian RNA Polymerase II at Site-specific Cisplatin Adducts in the Template DNA. J. Biol. Chem. 2003, 278, 35791–35797.
- Tremeau-Bravard, A.; Riedl, T.; Egly, J.-M.; Dahmus, M.E. Fate of RNA Polymerase II Stalled at a Cisplatin Lesion. J. Biol. Chem. 2004, 279, 7751–7759.
- Corda, Y.; Job, C.; Anin, M.F.; Leng, M.; Job, D. Transcription by eucaryotic and procaryotic RNA polymerases of DNA modified at a d(GG) or a d(AG) site by the antitumor drug cis-diamminedichloroplatinum(II). Biochemistry 1991, 30, 222–230.
- Corda, Y.; Anin, M.F.; Leng, M.; Job, D. RNA polymerases react differently at d(ApG) and d(GpG) adducts in DNA modified by cis-diamminedichloroplatinum(II). Biochemistry 1992, 31, 1904–1908.
- Corda, Y.; Job, C.; Anin, M.F.; Leng, M.; Job, D. Spectrum of DNA-platinum adduct recognition by prokaryotic and eukaryotic DNA-dependent RNA polymerases. Biochemistry 1993, 32, 8582–8588.
- Mello, J.A.; Lippard, S.J.; Essigmann, J.M. DNA Adducts of cis-Diamminedichloroplatinum(II) and Its Trans Isomer Inhibit RNA Polymerase II Differentially in Vivo. Biochemistry 1995, 34, 14783–14791.
- Todd, R.C.; Lippard, S.J. Inhibition of transcription by platinum antitumor compounds. Metallomics 2009, 1, 280–291.
- Vichi, P.; Coin, F.; Renaud, J.; Vermeulen, W.; Hoeijmakers, J.; Moras, D.; Egly, J. Cisplatin- and UV-damaged DNA lure the basal transcription factor TFIID/TBP. EMBO J. 1997, 16, 7444–7456.
- Cullinane, C.; Mazur, S.J.; Essigmann, J.M.; Phillips, A.D.R.; Bohr, V.A. Inhibition of RNA Polymerase II Transcription in Human Cell Extracts by Cisplatin DNA Damage. Biochemistry 1999, 38, 6204–6212.
- Treiber, D.K.; Zhai, X.; Jantzen, H.M.; Essigmann, J.M. Cisplatin-DNA adducts are molecular decoys for the ribosomal RNA transcription factor hUBF (human upstream binding factor). Proc. Natl. Acad. Sci. USA 1994, 91, 5672–5676.
- Zhai, X.; Beckmann, H.; Jantzen, H.-M.; Essigmann, J.M. Cisplatin−DNA Adducts Inhibit Ribosomal RNA Synthesis by Hijacking the Transcription Factor Human Upstream Binding Factor. Biochemistry 1998, 37, 16307–16315.
- Ise, T.; Nagatani, G.; Imamura, T.; Kato, K.; Takano, H.; Nomoto, M.; Izumi, H.; Ohmori, H.; Okamoto, T.; Ohga, T.; et al. Transcription factor Y-box binding protein 1 binds preferentially to cisplatin-modified DNA and interacts with proliferating cell nuclear antigen. Cancer Res. 1999, 59, 342–346.
- Yarnell, A.T.; Oh, S.; Reinberg, D.; Lippard, S.J. Interaction of FACT, SSRP1, and the High Mobility Group (HMG) Domain of SSRP1 with DNA Damaged by the Anticancer Drug Cisplatin *. J. Biol. Chem. 2001, 276, 25736–25741.
- Dunham, S.U.; Lippard, S.J. DNA Sequence Context and Protein Composition Modulate HMG-Domain Protein Recognition of Cisplatin-Modified DNA. Biochemistry 1997, 36, 11428–11436.
- Mymryk, J.S.; Zaniewski, E.; Archer, T.K. Cisplatin Inhibits Chromatin Remodeling, Transcription Factor Binding, and Transcription from the Mouse Mammary Tumor Virus Promoter in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 2076–2080.
- E Damsma, G.; Alt, A.; Brueckner, F.; Carell, T.; Cramer, P. Mechanism of transcriptional stalling at cisplatin-damaged DNA. Nat. Struct. Mol. Biol. 2007, 14, 1127–1133.
- Strauss, B.S. The ‘A rule’ of mutagen specificity: A consequence of DNA polymerase bypass of non-instructional lesions? BioEssays 1991, 13, 79–84.
- Wu, C. Chromatin Remodeling and the Control of Gene Expression. J. Biol. Chem. 1997, 272, 28171–28174.
- Li, B.; Carey, M.; Workman, J.L. The Role of Chromatin during Transcription. Cell 2007, 128, 707–719.
- Ober, M.; Lippard, S.J. Cisplatin Damage Overrides the Predefined Rotational Setting of Positioned Nucleosomes. J. Am. Chem. Soc. 2007, 129, 6278–6286.
- Ober, M.; Lippard, S.J. A 1,2-d(GpG) cisplatin intrastrand cross-link influences the rotational and translational setting of DNA in nucleosomes. J. Am. Chem. Soc. 2008, 130, 2851–2861.
- Todd, R.C.; Lippard, S.J. Consequences of Cisplatin Binding on Nucleosome Structure and Dynamics. Chem. Biol. 2010, 17, 1334–1343.
- Moon, H.-M.; Park, J.-S.; Lee, I.-B.; Kang, Y.-I.; Jung, H.J.; An, D.; Shin, Y.; Kim, M.J.; I Kim, H.; Song, J.-J.; et al. Cisplatin fastens chromatin irreversibly even at a high chloride concentration. Nucleic Acids Res. 2021, 49, 12035–12047.
- Charlier, C.; Kintz, P.; Dubois, N.; Plomteux, G. Fatal Overdosage with Cisplatin. J. Anal. Toxicol. 2004, 28, 138–140.
- Zhu, G.; Song, L.; Lippard, S.J. Visualizing Inhibition of Nucleosome Mobility and Transcription by Cisplatin–DNA Interstrand Crosslinks in Live Mammalian Cells. Cancer Res. 2013, 73, 4451–4460.
- Pucci, B.; Kasten, M.; Giordano, A. Cell cycle and apoptosis. Neoplasia 2000, 2, 291–299.
- Donahue, B.A.; Augot, M.; Bellon, S.F.; Treiber, D.K.; Toney, J.H.; Lippard, S.J.; Essigmann, J.M. Characterization of a DNA damage-recognition protein from mammalian cells that binds specifically to intrastrand d(GpG) and d(ApG) DNA adducts of the anticancer drug cisplatin. Biochemistry 1990, 29, 5872–5880.
- Fink, D.; Aebi, S.; Howell, S.B. The role of DNA mismatch repair in drug resistance. Clin. Cancer Res. 1998, 4, 1–6.
- Chaney, S.G.; Vaisman, A. Specificity of platinum–DNA adduct repair. J. Inorg. Biochem. 1999, 77, 71–81.
- Appella, E.; Anderson, C.W. Post-translational modifications and activation of p53 by genotoxic stresses. JBIC J. Biol. Inorg. Chem. 2001, 268, 2764–2772.
- Persons, D.L.; Yazlovitskaya, E.M.; Pelling, J.C. Effect of Extracellular Signal-regulated Kinase on p53 Accumulation in Response to Cisplatin. J. Biol. Chem. 2000, 275, 35778–35785.
- Singh, M.; Chaudhry, P.; Fabi, F.; Asselin, E. Cisplatin-induced caspase activation mediates PTEN cleavage in ovarian cancer cells: A potential mechanism of chemoresistance. BMC Cancer 2013, 13, 233–239.
- Cummings, B.S.; Schnellmann, R.G. Cisplatin-Induced Renal Cell Apoptosis: Caspase 3-Dependent and -Independent Pathways. J. Pharmacol. Exp. Ther. 2002, 302, 8–17.
- Jones, E.V.; Dickman, M.; Whitmarsh, A.J. Regulation of p73-mediated apoptosis by c-Jun N-terminal kinase. Biochem. J. 2007, 405, 617–623.
- Paul, I.; Chacko, A.D.; Stasik, I.; Busacca, S.; Crawford, N.; McCoy, F.; McTavish, N.; Wilson, B.; Barr, M.; O’Byrne, K.J.; et al. Acquired differential regulation of caspase-8 in cisplatin-resistant non-small-cell lung cancer. Cell Death Dis. 2012, 3, e449.
- Mansouri, A.; Ridgway, L.D.; Korapati, A.L.; Zhang, Q.; Tian, L.; Wang, Y.; Siddik, Z.H.; Mills, G.B.; Claret, F.X. Sustained Activation of JNK/p38 MAPK Pathways in Response to Cisplatin Leads to Fas Ligand Induction and Cell Death in Ovarian Carcinoma Cells. J. Biol. Chem. 2003, 278, 19245–19256.
- Yang, Z.; Schumaker, L.M.; Egorin, M.J.; Zuhowski, E.G.; Guo, Z.; Cullen, K.J. Cisplatin Preferentially Binds Mitochondrial DNA and Voltage-Dependent Anion Channel Protein in the Mitochondrial Membrane of Head and Neck Squamous Cell Carcinoma: Possible Role in Apoptosis. Clin. Cancer Res. 2006, 12, 5817–5825.
- Mandic, A.; Hansson, J.; Linder, S.; Shoshan, M.C. Cisplatin Induces Endoplasmic Reticulum Stress and Nucleus-independent Apoptotic Signaling. J. Biol. Chem. 2003, 278, 9100–9106.
- Yu, F.; Megyesi, J.; Price, P.M. Cytoplasmic initiation of cisplatin cytotoxicity. Am. J. Physiol. -Renal Physiol. 2008, 295, F44–F52.
- Gutekunst, M.; Oren, M.; Weilbacher, A.; Dengler, M.A.; Markwardt, C.; Thomale, J.; Aulitzky, W.E.; Van Der Kuip, H. p53 Hypersensitivity Is the Predominant Mechanism of the Unique Responsiveness of Testicular Germ Cell Tumor (TGCT) Cells to Cisplatin. PLoS ONE 2011, 6, e19198.
- Olivero, O.A.; Chang, P.K.; Lopez-Larraza, D.M.; Semino-Mora, M.C.; Poirier, M.C. Preferential formation and decreased removal of cisplatin–DNA adducts in Chinese hamster ovary cell mitochondrial DNA as compared to nuclear DNA. Mutat. Res. Toxicol. Environ. Mutagen. 1997, 391, 79–86.
- Montopoli, M.; Bellanda, M.; Lonardoni, F.; Ragazzi, E.; Dorigo, P.; Froldi, G.; Mammi, S.; Caparrotta, L. “Metabolic reprogramming” in ovarian cancer cells resistant to cisplatin. Curr. Cancer Drug Targets 2011, 11, 226–235.
- Martins, N.M.; Santos, N.A.G.; Curti, C.; Bianchi, M.L.P.; Santos, A.C. Cisplatin induces mitochondrial oxidative stress with resultant energetic metabolism impairment, membrane rigidification and apoptosis in rat liver. J. Appl. Toxicol. 2007, 28, 337–344.
- Santandreu, F.M.; Roca, P.; Oliver, J. Uncoupling protein-2 knockdown mediates the cytotoxic effects of cisplatin. Free Radic. Biol. Med. 2010, 49, 658–666.
- Li-ping, X.; Skrezek, C.; Wand, H.; Reibe, F. Mitochondrial dysfunction at the early stage of cisplatin-induced acute renal failure in rats. J. Zhejiang Univ. -Sci. 2000, 1, 91–96.
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin Induces a Mitochondrial-ROS Response That Contributes to Cytotoxicity Depending on Mitochondrial Redox Status and Bioenergetic Functions. PLoS ONE 2013, 8, e81162.
- Choi, Y.-M.; Kim, H.; Shim, W.; Anwar, M.A.; Kwon, J.-W.; Kwon, H.-K.; Kim, H.J.; Jeong, H.; Kim, H.M.; Hwang, D.; et al. Mechanism of Cisplatin-Induced Cytotoxicity Is Correlated to Impaired Metabolism Due to Mitochondrial ROS Generation. PLoS ONE 2015, 10, e0135083.
- Garrido, N.; Pérez-Martos, A.; Faro, M.; Lou-Bonafonte, J.M.; Fernández-Silva, P.; López-Pérez, M.J.; Montoya, J.; Enríquez, J.A. Cisplatin-mediated impairment of mitochondrial DNA metabolism inversely correlates with glutathione levels. Biochem. J. 2008, 414, 93–102.
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2016, 1863, 2977–2992.
- Fleury, C.; Mignotte, B.; Vayssière, J.-L. Mitochondrial reactive oxygen species in cell death signaling. Biochimie 2002, 84, 131–141.
- Wu, C.-C.; Bratton, S.B. Regulation of the Intrinsic Apoptosis Pathway by Reactive Oxygen Species. Antioxidants Redox Signal. 2013, 19, 546–558.
- Halestrap, A.P.; Woodfield, K.-Y.; Connern, C.P. Oxidative Stress, Thiol Reagents, and Membrane Potential Modulate the Mitochondrial Permeability Transition by Affecting Nucleotide Binding to the Adenine Nucleotide Translocase *. J. Biol. Chem. 1997, 272, 3346–3354.
- Kagan, V.E.; Borisenko, G.G.; Tyurina, Y.Y.; Tyurin, V.; Jiang, J.; Potapovich, A.I.; Kini, V.; Amoscato, A.A.; Fujii, Y. Oxidative lipidomics of apoptosis: Redox catalytic interactions of cytochrome c with cardiolipin and phosphatidylserine. Free. Radic. Biol. Med. 2004, 37, 1963–1985.
- Kleih, M.; Böpple, K.; Dong, M.; Gaißler, A.; Heine, S.; Olayioye, M.A.; Aulitzky, W.E.; Essmann, F. Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis. 2019, 10, 851.
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94.
- Avril, T.; Vauléon, E.; Chevet, E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 2017, 6, e373.
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102.
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381.
- Costa-Mattioli, M.; Walter, P. The integrated stress response: From mechanism to disease. Science 2020, 368.
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190.
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2019, 9, 3083.
- Yoneda, T.; Imaizumi, K.; Oono, K.; Yui, D.; Gomi, F.; Katayama, T.; Tohyama, M. Activation of Caspase-12, an Endoplastic Reticulum (ER) Resident Caspase, through Tumor Necrosis Factor Receptor-associated Factor 2-dependent Mechanism in Response to the ER Stress *. J. Biol. Chem. 2001, 276, 13935–13940.
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 2000, 403, 98–103.
- Yang, H.; Niemeijer, M.; van de Water, B.; Beltman, J.B. ATF6 Is a Critical Determinant of CHOP Dynamics during the Unfolded Protein Response. iScience 2020, 23, 100860.
- Xu, Y.; Yu, H.; Qin, H.; Kang, J.; Yu, C.; Zhong, J.; Su, J.; Li, H.; Sun, L. Inhibition of autophagy enhances cisplatin cytotoxicity through endoplasmic reticulum stress in human cervical cancer cells. Cancer Lett. 2012, 314, 232–243.
- Xu, Y.; Wang, C.; Su, J.; Xie, Q.; Ma, L.; Zeng, L.; Yu, Y.; Liu, S.; Li, S.; Li, Z.; et al. Tolerance to endoplasmic reticulum stress mediates cisplatin resistance in human ovarian cancer cells by maintaining endoplasmic reticulum and mitochondrial homeostasis. Oncol. Rep. 2015, 34, 3051–3060.
- Liu, H.; Baliga, R. Endoplasmic Reticulum Stress–Associated Caspase 12 Mediates Cisplatin-Induced LLC-PK1 Cell Apoptosis. J. Am. Soc. Nephrol. 2005, 16, 1985–1992.
- Shen, L.; Wen, N.; Xia, M.; Zhang, Y.; Liu, W.; Xu, Y.; Sun, L. Calcium efflux from the endoplasmic reticulum regulates cisplatin-induced apoptosis in human cervical cancer HeLa cells. Oncol. Lett. 2016, 11, 2411–2419.
- Li, C.; Wei, J.; Li, Y.; He, X.; Zhou, Q.; Yan, J.; Zhang, J.; Liu, Y.; Shu, H.-B. Transmembrane Protein 214 (TMEM214) Mediates Endoplasmic Reticulum Stress-induced Caspase 4 Enzyme Activation and Apoptosis. J. Biol. Chem. 2013, 288, 17908–17917.
- Harwood, S.M.; Yaqoob, M.M.; A Allen, D. Caspase and calpain function in cell death: Bridging the gap between apoptosis and necrosis. Ann. Clin. Biochem. Int. J. Biochem. Lab. Med. 2005, 42, 415–431.
- Danese, A.; Leo, S.; Rimessi, A.; Wieckowski, M.R.; Fiorica, F.; Giorgi, C.; Pinton, P. Cell death as a result of calcium signaling modulation: A cancer-centric prospective. Biochim. et Biophys. Acta (BBA) Mol. Cell Res. 2021, 1868, 119061.
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730.
- Akman, M.; Belisario, D.C.; Salaroglio, I.C.; Kopecka, J.; Donadelli, M.; De Smaele, E.; Riganti, C. Hypoxia, endoplasmic reticulum stress and chemoresistance: Dangerous liaisons. J. Exp. Clin. Cancer Res. 2021, 40, 1–17.
- Tian, J.; Liu, R.; Qu, Q. Role of endoplasmic reticulum stress on cisplatin resistance in ovarian carcinoma. Oncol. Lett. 2017, 13, 1437–1443.
- Rashid, H.-O.; Yadav, R.K.; Kim, H.-R.; Chae, H.-J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977.
- Burger, K.N.; Staffhorst, R.W.; De Kruijff, B. Interaction of the anti-cancer drug cisplatin with phosphatidylserine in intact and semi-intact cells. Biochim. Biophys. Acta (BBA) -Biomembr. 1999, 1419, 43–54.
- Speelmans, G.; Sips, W.H.; Grisel, R.J.; Staffhorst, R.W.; Fichtinger-Schepman, A.M.J.; Reedijk, J.; de Kruijff, B. The interaction of the anti-cancer drug cisplatin with phospholipids is specific for negatively charged phospholipids and takes place at low chloride ion concentration. Biochim. Biophys. Acta (BBA) -Biomembr. 1996, 1283, 60–66.
- Suwalsky, M.; Hernández, P.; Villena, F.; Sotomayor, C.P. The Anticancer Drug Cisplatin Interacts with the Human Erythrocyte Membrane. Z. Naturforschung C 2000, 55, 461–466.
- Ramachandran, S.; Quist, A.P.; Kumar, S.; Lal, R. Cisplatin Nanoliposomes for Cancer Therapy: AFM and Fluorescence Imaging of Cisplatin Encapsulation, Stability, Cellular Uptake, and Toxicity. Langmuir 2006, 22, 8156–8162.
- Wang, K.; Lu, J.; Li, R. The events that occur when cisplatin encounters cells. Coord. Chem. Rev. 1996, 151, 53–88.
- Lacour, S.; Hammann, A.; Grazide, S.; Lagadic-Gossmann, D.; Athias, A.; Sergent, O.; Laurent, G.; Gambert, P.; Solary, E.; Dimanche-Boitrel, M.-T. Cisplatin-Induced CD95 Redistribution into Membrane Lipid Rafts of HT29 Human Colon Cancer Cells. Cancer Res. 2004, 64, 3593–3598.
- Rebillard, A.; Tekpli, X.; Meurette, O.; Sergent, O.; LeMoigne-Muller, G.; Vernhet, L.; Gorria, M.; Chevanne, M.; Christmann, M.; Kaina, B.; et al. Cisplatin-Induced Apoptosis Involves Membrane Fluidification via Inhibition of NHE1 in Human Colon Cancer Cells. Cancer Res. 2007, 67, 7865–7874.
- Zhang, Y.; Zeng, W.; Jia, F.; Ye, J.; Zhao, Y.; Luo, Q.; Zhu, Z.; Wang, F. Cisplatin-induced alteration on membrane composition of A549 cells revealed by ToF-SIMS. Surf. Interface Anal. 2020, 52, 256–263.
- Nganga, R.; Oleinik, N.; Ogretmen, B. Chapter One—Mechanisms of Ceramide-Dependent Cancer Cell Death. In Advances in Cancer Research; Chalfant, C.E., Fisher, P.B., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 140, pp. 1–25.
- Fife, C.M.; A McCarroll, J.; Kavallaris, M. Movers and shakers: Cell cytoskeleton in cancer metastasis. Br. J. Pharmacol. 2014, 171, 5507–5523.
- Köpf-Maier, P.; Mühlhausen, S. Changes in the cytoskeleton pattern of tumor cells by cisplatin in vitro. Chem. Interact. 1992, 82, 295–316.
- Zeidan, Y.H.; Jenkins, R.W.; Hannun, Y.A. Remodeling of cellular cytoskeleton by the acid sphingomyelinase/ceramide pathway. J. Cell Biol. 2008, 181, 335–350.
- Min, Y.J.; Poruchynsky, M.S.; Sackett, D.L.; Murphy, B.; Fojo, T. Cisplatin markedly enhances microtubule depolymerization in A549 cell line compared with oxaliplatin. Cancer Res. 2008, 68 (Suppl. 9), 3335.
- Tulub, A.A.; Stefanov, V.E. Cisplatin stops tubulin assembly into microtubules. A new insight into the mechanism of antitumor activity of platinum complexes. Int. J. Biol. Macromol. 2001, 28, 191–198.
- Raudenska, M.; Kratochvilova, M.; Vicar, T.; Gumulec, J.; Balvan, J.; Polanska, H.; Pribyl, J.; Masarik, M. Cisplatin enhances cell stiffness and decreases invasiveness rate in prostate cancer cells by actin accumulation. Sci. Rep. 2019, 9, 1660.
- Pierson-Marchandise, M.; Gras, V.; Moragny, J.; Micallef, J.; Gaboriau, L.; Picard, S.; Choukroun, G.; Masmoudi, K.; Liabeuf, S.; The French National Network of Pharmacovigilance Centres. The drugs that mostly frequently induce acute kidney injury: A case − noncase study of a pharmacovigilance database. Br. J. Clin. Pharmacol. 2017, 83, 1341–1349.
- Hoek, J.; Bloemendal, K.M.; van der Velden, L.A.; van Diessen, J.N.; van Werkhoven, E.; Klop, W.M.; Tesselaar, M.E. Nephrotoxicity as a Dose-Limiting Factor in a High-Dose Cisplatin-Based Chemoradiotherapy Regimen for Head and Neck Carcinomas. Cancers 2016, 8, 21.
- Hanigan, M.H.; Devarajan, P. Cisplatin nephrotoxicity: Molecular mechanisms. Cancer Ther. 2003, 1, 47–61.
- Gonzalez-Vitale, J.C.; Hayes, D.M.; Cvitkovic, E.; Sternberg, S.S. The renal pathology in clinical trials of Cis-platinum (II) diamminedichloride. Cancer 1977, 39, 1362–1371.
- Arany, I.; Safirstein, R.L. Cisplatin nephrotoxicity. Semin. Nephrol. 2003, 23, 460–464.
- Megyesi, J.; Safirstein, R.L.; Price, P.M. Induction of p21WAF1/CIP1/SDI1 in kidney tubule cells affects the course of cisplatin-induced acute renal failure. J. Clin. Investig. 1998, 101, 777–782.
- Tsuruya, K.; Ninomiya, T.; Tokumoto, M.; Hirakawa, M.; Masutani, K.; Taniguchi, M.; Fukuda, K.; Kanai, H.; Kishihara, K.; Hirakata, H.; et al. Direct involvement of the receptor-mediated apoptotic pathways in cisplatin-induced renal tubular cell death. Kidney Int. 2003, 63, 72–82.
- Baliga, R.; Ueda, N.; Walker, P.D.; Shah, S.V. Oxidant Mechanisms in Toxic Acute Renal Failure*. Drug Metab. Rev. 1999, 31, 971–997.
- Ramesh, G.; Reeves, W.B. Cisplatin Increases TNF-α mRNA Stability in Kidney Proximal Tubule Cells. Ren. Fail. 2006, 28, 583–592.
- Ramesh, G.; Reeves, W.B. TNF-α mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J. Clin. Investig. 2002, 110, 835–842.
- Ramesh, G.; Reeves, W. TNFR2-mediated apoptosis and necrosis in cisplatin-induced acute renal failure. Am. J. Physiol. Physiol. 2003, 285, F610–F618.
- Winston, J.A.; Safirstein, R. Reduced renal blood flow in early cisplatin-induced acute renal failure in the rat. Am. J. Physiol. Physiol. 1985, 249, F490–F496.
- Offerman, J.J.G.; Meijer, S.; Sleijfer, D.T.; Mulder, N.H.; Donker, A.J.M.; Koops, H.S.; Van Der Hem, G.K. Acute effects of cis-diamminedichloroplatinum (CDDP) on renal function. Cancer Chemother. Pharmacol. 1984, 12, 36–38.
- Callejo, A.; Sedó-Cabezón, L.; Juan, I.D.; Llorens, J. Cisplatin-Induced Ototoxicity: Effects, Mechanisms and Protection Strategies. Toxics 2015, 3, 268–293.
- Knight, K.R.G.; Kraemer, D.F.; Neuwelt, E.A. Ototoxicity in Children Receiving Platinum Chemotherapy: Underestimating a Commonly Occurring Toxicity That May Influence Academic and Social Development. J. Clin. Oncol. 2005, 23, 8588–8596.
- Kushner, B.H.; Budnick, A.; Kramer, K.; Modak, S.; Cheung, N.-K.V. Ototoxicity from high-dose use of platinum compounds in patients with neuroblastoma. Cancer 2006, 107, 417–422.
- Kopke, R.; Allen, K.A.; Henderson, D.; Hoffer, M.; Frenz, D.; VAN DE Water, T. A Radical Demise: Toxins and Trauma Share Common Pathways in Hair Cell Death. Ann. N. Y. Acad. Sci. 1999, 884, 171–191.
- Breglio, A.M.; Rusheen, A.E.; Shide, E.D.; Fernandez, K.A.; Spielbauer, K.K.; McLachlin, K.M.; Hall, M.D.; Amable, L.; Cunningham, L.L. Cisplatin is retained in the cochlea indefinitely following chemotherapy. Nat. Commun. 2017, 8, 1654.
- Filipski, K.K.; Mathijssen, R.H.; Mikkelsen, T.S.; Schinkel, A.H.; Sparreboom, A. Contribution of Organic Cation Transporter 2 (OCT2) to Cisplatin-Induced Nephrotoxicity. Clin. Pharmacol. Ther. 2009, 86, 396–402.
- Ciarimboli, G.; Deuster, D.; Knief, A.; Sperling, M.; Holtkamp, M.; Edemir, B.; Pavenstädt, H.; Lanvers-Kaminsky, C.; am Zehnhoff-Dinnesen, A.; Schinkel, A.H.; et al. Organic cation transporter 2 mediates cisplatin-induced oto- and nephrotoxicity and is a target for protective interventions. Am. J. Pathol. 2010, 176, 1169–1180.
- Gregg, R.W.; Molepo, J.M.; Monpetit, V.J.; Mikael, N.Z.; Redmond, D.; Gadia, M.; Stewart, D.J. Cisplatin neurotoxicity: The relationship between dosage, time, and platinum concentration in neurologic tissues, and morphologic evidence of toxicity. J. Clin. Oncol. 1992, 10, 795–803.
- Starobova, H.; Vetter, I. Pathophysiology of Chemotherapy-Induced Peripheral Neuropathy. Front. Mol. Neurosci. 2017, 10, 174.
- Lomonaco, M.; Milone, M.; Batocchi, A.P.; Padua, L.; Restuccia, D.; Tonali, P. Cisplatin neuropathy: Clinical course and neurophysiological findings. J. Neurol. 1992, 239, 199–204.
- Siegal, T.; Haim, N. Cisplatin-induced peripheral neuropathy. Frequent off-therapy deterioration, demyelinating syndromes, and muscle cramps. Cancer 1990, 66, 1117–1123.
- Kerckhove, N.; Collin, A.; Condé, S.; Chaleteix, C.; Pezet, D.; Balayssac, D. Long-Term Effects, Pathophysiological Mechanisms, and Risk Factors of Chemotherapy-Induced Peripheral Neuropathies: A Comprehensive Literature Review. Front. Pharmacol. 2017, 8, 86.
- Cavalli, F.; Tschopp, L.; Sonntag, R.W.; Zimmermann, A. A case of liver toxicity following cis-dichlorodiammineplatinum(II) treatment. Cancer Treat. Rep. 1978, 62, 2125–2126.
- Hu, Y.; Sun, B.; Zhao, B.; Mei, D.; Gu, Q.; Tian, Z. Cisplatin-induced cardiotoxicity with midrange ejection fraction: A case report and review of the literature. Medicine 2018, 97, e13807.
- Astolfi, L.; Ghiselli, S.; Guaran, V.; Chicca, M.; Simoni, E.; Olivetto, E.; Lelli, G.; Martini, A. Correlation of adverse effects of cisplatin administration in patients affected by solid tumours: A retrospective evaluation. Oncol. Rep. 2013, 29, 1285–1292.
- Batchelor, D. Hair and cancer chemotherapy: Consequences and nursing care—A literature study. Eur. J. Cancer Care 2001, 10, 147–163.
- Kartalou, M.; Essigmann, J.M. Mechanisms of resistance to cisplatin. Mutation Res. /Fundam. Molecular Mech. Mutagen. 2001, 478, 23–43.
- Kilari, D.; Guancial, E.; Kim, E.S. Role of copper transporters in platinum resistance. World J. Clin. Oncol. 2016, 7, 106–113.
- Öhrvik, H.; Logeman, B.; Turk, B.; Reinheckel, T.; Thiele, D.J. Cathepsin Protease Controls Copper and Cisplatin Accumulation via Cleavage of the Ctr1 Metal-binding Ectodomain. J. Biol. Chem. 2016, 291, 13905–13916.
- Inoue, Y.; Matsumoto, H.; Yamada, S.; Kawai, K.; Suemizu, H.; Gika, M.; Takanami, I.; Iwazaki, M.; Nakamura, M. Association of ATP7A expression and in vitro sensitivity to cisplatin in non-small cell lung cancer. Oncol. Lett. 2010, 1, 837–840.
- Samimi, G.; Varki, N.M.; Wilczynski, S.; Safaei, R.; Alberts, D.S.; Howell, S.B. Increase in expression of the copper transporter ATP7A during platinum drug-based treatment is associated with poor survival in ovarian cancer patients. Clin. Cancer Res. 2003, 9, 5853–5859.
- Yang, T.; Chen, M.; Chen, T.; Thakur, A. Expression of the copper transporters hCtr1, ATP7A and ATP7B is associated with the response to chemotherapy and survival time in patients with resected non-small cell lung cancer. Oncol. Lett. 2015, 10, 2584–2590.
- Song, L.; Li, Y.; Li, W.; Wu, S.; Li, Z. miR-495 Enhances the Sensitivity of Non-Small Cell Lung Cancer Cells to Platinum by Modulation of Copper-Transporting P-type Adenosine Triphosphatase A (ATP7A). J. Cell. Biochem. 2013, 115, 1234–1242.
- Yu, Z.; Cao, W.; Ren, Y.; Zhang, Q.; Liu, J. ATPase copper transporter A, negatively regulated by miR-148a-3p, contributes to cisplatin resistance in breast cancer cells. Clin. Transl. Med. 2020, 10, 57–73.
- Hinoshita, E.; Uchiumi, T.; Taguchi, K.; Kinukawa, N.; Tsuneyoshi, M.; Maehara, Y.; Sugimachi, K.; Kuwano, M. Increased expression of an ATP-binding cassette superfamily transporter, multidrug resistance protein 2, in human colorectal carcinomas. Clin. Cancer Res. 2000, 6, 2401–2407.
- Yamasaki, M.; Makino, T.; Masuzawa, T.; Kurokawa, Y.; Miyata, H.; Takiguchi, S.; Nakajima, K.; Fujiwara, Y.; Matsuura, N.; Mori, M.; et al. Role of multidrug resistance protein 2 (MRP2) in chemoresistance and clinical outcome in oesophageal squamous cell carcinoma. Br. J. Cancer 2011, 104, 707–713.
- Amable, L. Cisplatin resistance and opportunities for precision medicine. Pharmacol. Res. 2016, 106, 27–36.
- Rudin, C.M.; Yang, Z.; Schumaker, L.M.; VanderWeele, D.; Newkirk, K.; Egorin, M.J.; Zuhowski, E.G.; Cullen, K.J. Inhibition of glutathione synthesis reverses Bcl-2-mediated cisplatin resistance. Cancer Res. 2003, 63, 312–318.
- Chen, H.H.W.; Kuo, M.T. Role of Glutathione in the Regulation of Cisplatin Resistance in Cancer Chemotherapy. Met. Based Drugs 2010, 2010, 430939.
- Byun, S.-S.; Kim, S.W.; Choi, H.; Lee, C.; Lee, E. Augmentation of cisplatin sensitivity in cisplatin-resistant human bladder cancer cells by modulating glutathione concentrations and glutathione-related enzyme activities. BJU Int. 2005, 95, 1086–1090.
- Rocha, C.R.R.; Garcia, C.C.M.; Vieira, D.B.; Quinet, A.; de Andrade-Lima, L.C.; Munford, V.; Belizário, J.E.; Menck, C.F.M. Glutathione depletion sensitizes cisplatin- and temozolomide-resistant glioma cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1505.
- Hayden, A.; Douglas, J.; Sommerlad, M.; Andrews, L.; Gould, K.; Hussain, S.; Thomas, G.J.; Packham, G.; Crabb, S.J. The Nrf2 transcription factor contributes to resistance to cisplatin in bladder cancer. Urol. Oncol. Semin. Orig. Investig. 2014, 32, 806–814.
- Li, D.; Hong, X.; Zhao, F.; Ci, X.; Zhang, S. Targeting Nrf2 may reverse the drug resistance in ovarian cancer. Cancer Cell Int. 2021, 21, 1–10.
- Wangy, X.-J.; Suny, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinog. 2008, 29, 1235–1243.
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88 Pt B, 199–204.
- Yu, H.; Su, J.; Xu, Y.; Kang, J.; Li, H.; Zhang, L.; Yi, H.; Xiang, X.; Liu, F.; Sun, L. p62/SQSTM1 involved in cisplatin resistance in human ovarian cancer cells by clearing ubiquitinated proteins. Eur. J. Cancer 2011, 47, 1585–1594.
- Bao, L.-J.; Jaramillo, M.C.; Zhang, Z.-B.; Zheng, Y.-X.; Yao, M.; Zhang, D.D.; Yi, X.-F. Nrf2 induces cisplatin resistance through activation of autophagy in ovarian carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 1502–1513.
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s.
- Schärer, O.D. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609.
- Duan, M.; Ulibarri, J.; Liu, K.J.; Mao, P. Role of Nucleotide Excision Repair in Cisplatin Resistance. Int. J. Mol. Sci. 2020, 21, 9248.
- Ng, J.M.; Vermeulen, W.; van der Horst, G.T.; Bergink, S.; Sugasawa, K.; Vrieling, H.; Hoeijmakers, J.H. A novel regulation mechanism of DNA repair by damage-induced and RAD23-dependent stabilization of xeroderma pigmentosum group C protein. Genes Dev. 2003, 17, 1630–1645.
- Nocentini, S.; Coin, F.; Saijo, M.; Tanaka, K.; Egly, J.-M. DNA Damage Recognition by XPA Protein Promotes Efficient Recruitment of Transcription Factor II H. J. Biol. Chem. 1997, 272, 22991–22994.
- Yokoi, M.; Masutani, C.; Maekawa, T.; Sugasawa, K.; Ohkuma, Y.; Hanaoka, F. The Xeroderma Pigmentosum Group C Protein Complex XPC-HR23B Plays an Important Role in the Recruitment of Transcription Factor IIH to Damaged DNA. J. Biol. Chem. 2000, 275, 9870–9875.
- Oksenych, V.; de Jesus, B.B.; Zhovmer, A.; Egly, J.-M.; Coin, F. Molecular insights into the recruitment of TFIIH to sites of DNA damage. EMBO J. 2009, 28, 2971–2980.
- Coin, F.; Oksenych, V.; Egly, J.-M. Distinct Roles for the XPB/p52 and XPD/p44 Subcomplexes of TFIIH in Damaged DNA Opening during Nucleotide Excision Repair. Mol. Cell 2007, 26, 245–256.
- Houtsmuller, A.B.; Rademakers, S.; Nigg, A.L.; Hoogstraten, D.; Hoeijmakers, J.H.; Vermeulen, W. Action of DNA repair endonuclease ERCC1/XPF in living cells. Science 1999, 284, 958–961.
- Ito, S.; Kuraoka, I.; Chymkowitch, P.; Compe, E.; Takedachi, A.; Ishigami, C.; Coin, F.; Egly, J.-M.; Tanaka, K. XPG Stabilizes TFIIH, Allowing Transactivation of Nuclear Receptors: Implications for Cockayne Syndrome in XP-G/CS Patients. Mol. Cell 2007, 26, 231–243.
- Kemp, M.G.; Reardon, J.T.; Lindsey-Boltz, L.; Sancar, A. Mechanism of Release and Fate of Excised Oligonucleotides during Nucleotide Excision Repair. J. Biol. Chem. 2012, 287, 22889–22899.
- Ogi, T.; Limsirichaikul, S.; Overmeer, R.M.; Volker, M.; Takenaka, K.; Cloney, R.; Nakazawa, Y.; Niimi, A.; Miki, Y.; Jaspers, N.G.; et al. Three DNA Polymerases, Recruited by Different Mechanisms, Carry Out NER Repair Synthesis in Human Cells. Mol. Cell 2010, 37, 714–727.
- Ferry, K.V.; Hamilton, T.C.; Johnson, S.W. Increased nucleotide excision repair in cisplatin-resistant ovarian cancer cells: Role of ercc1–xpf. Biochem. Pharmacol. 2000, 60, 1305–1313.
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous Recombination and Human Health: The Roles of BRCA1, BRCA2, and Associated Proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600.
- Toh, M.; Ngeow, J. Homologous Recombination Deficiency: Cancer Predispositions and Treatment Implications. Oncologist 2021, 26, e1526–e1537.
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120.
- Swisher, E.M.; Sakai, W.; Karlan, B.Y.; Wurz, K.; Urban, N.; Taniguchi, T. Secondary BRCA1 Mutations in BRCA1-Mutated Ovarian Carcinomas with Platinum Resistance. Cancer Res. 2008, 68, 2581–2586.
- Sawant, A.; Kothandapani, A.; Zhitkovich, A.; Sobol, R.W.; Patrick, S.M. Role of mismatch repair proteins in the processing of cisplatin interstrand cross-links. DNA Repair 2015, 35, 126–136.
- Yamada, M.; O’Regan, E.; Brown, R.; Karran, P. Selective recognition of a cisplatin-DNA adduct by human mismatch repair proteins. Nucleic Acids Res. 1997, 25, 491–496.
- Stojic, L.; Brun, R.; Jiricny, J. Mismatch repair and DNA damage signalling. DNA Repair 2004, 3, 1091–1101.
- Topping, R.P.; Wilkinson, J.C.; Scarpinato, K.D. Mismatch Repair Protein Deficiency Compromises Cisplatin-induced Apoptotic Signaling. J. Biol. Chem. 2009, 284, 14029–14039.
- Aebi, S.; Kurdi-Haidar, B.; Gordon, R.; Cenni, B.; Zheng, H.; Fink, D.; Christen, R.D.; Boland, C.R.; Koi, M.; Fishel, R.; et al. Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer Res. 1996, 56, 3087–3090.
- Fink, D.; Zheng, H.; Nebel, S.; Norris, P.S.; Aebi, S.; Lin, T.P.; Nehmé, A.; Christen, R.D.; Haas, M.; MacLeod, C.L.; et al. In vitro and in vivo resistance to cisplatin in cells that have lost DNA mismatch repair. Cancer Res. 1997, 57, 1841–1845.
- Fink, D.; Nebel, S.; Norris, P.S.; Baergen, R.N.; Wilczynski, S.P.; Costa, M.J.; Haas, M.; Cannistra, S.A.; Howell, S.B. Enrichment for DNA mismatch repair-deficient cells during treatment with cisplatin. Int. J. Cancer 1998, 77, 741–746.