1. Clinical Usage of Cisplatin
CDDP, despite being approved over 40 years ago, is still used today against many cancers. These include breast, ovarian, testicular, head and neck, esophageal, lung, bladder, and brain tumours [
23] (
Table 1).
Table 1. Comparison of indications and major side effects of cisplatin, carboplatin, and oxaliplatin.
The rise in the use of CDDP for treatment of these cancers resulted in a marked increase in survival, with testicular cancer as a particularly remarkable example. Prior to CDDP treatment, disease-free survival rates for testicular cancer ranged from 5 to 10%; however, with the introduction of CDDP in the 1980s, these rates drastically increased to 85% [
24]. Today, testicular cancer is virtually curable in the early stages, with the disease-free survival hovering at 95% [
25]. CDDP, in addition to working well as a cancer therapeutic on its own, also contributed to the practice of combination chemotherapy, becoming a base upon which, many mixed therapies were developed [
9]. CDDP is currently approved in combination with over twenty other anti-cancer drugs for the treatment of a variety of cancers, and hundreds of trials are currently being performed using CDDP in combination with other drugs [
9,
26].
2. Cellular Uptake of Cisplatin
CDDP is most commonly administered as cycles of IV injection given every three to four weeks. In the blood, roughly 90% of CDDP is sequestered by plasma proteins, leaving just 10% to access tissues [
27]. The mechanism of CDDP uptake into cells is currently a subject of debate. Initially, it was thought that CDDP entered the cell solely through passive diffusion due to its relatively small size and neutral charge. Indeed, the ability of CDDP to diffuse through lipid bilayers has been shown extensively in vitro [
28,
29,
30]. In addition, key pieces of evidence support the notion of a mainly passive mode of CDDP transport (
Figure 2). First, it has been demonstrated that intracellular CDDP uptake remains unchanged upon co-treatment with analogs [
31,
32]. Human ovarian carcinoma cells were treated with CDDP along with excess carboplatin, transplatin, and cis-PdCl
2(NH
3)
2. The presence of these structural analogs was unable to inhibit CDDP uptake, even when using concentrations 60-fold higher than that of CDDP in the case of carboplatin [
31]. Cis-PdCl
2(NH
3)
2 was the only compound capable of decreasing CDDP uptake, reducing accumulation by 20%. However, the authors suggest that this decrease was likely due to nonspecific damage from the reactive palladium compound, rather than to inhibition of a transport protein which would have had an expected decrease of 70% [
31]. Additional support for passive diffusion comes from a 1973 study in which Gale et al. found that accumulation of cis-diammine(dipyridine) Pt (II) could not be saturated in Ehrlich ascites tumour cells. Indeed, the rate of uptake remained linear up until the compound reached its saturation limit in DMSO [
28]. Since then, this finding has been confirmed by others using CDDP in other cell lines [
33,
34].
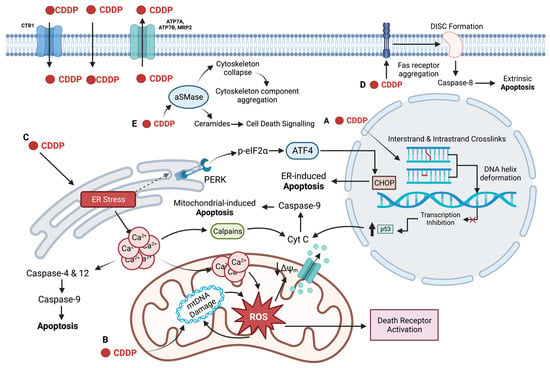
Figure 2. CDDP targets the nucleus, mitochondria, ER, plasma membrane, and cytoskeleton to induce cell death in its role as an anti-cancer drug. CDDP enters the cell via passive diffusion and high affinity copper uptake protein 1 (CTR1) and is removed from the cell by efflux transporters, ATP7A, ATP7B, and multidrug resistance protein 2 (MRP2). (
A) CDDP forms intrastrand and interstrand crosslinks, distorting the DNA helix, inhibiting transcription (represented by the red cross) and activating p53-mediated apoptosis. (
B) CDDP damages mitochondrial DNA (mtDNA), preventing transcription of electron transport chain (ETC) genes. Impaired ETC function results in ROS formation that further damages the mitochondria and activates the intrinsic and extrinsic apoptotic pathways. (
C) CDDP induces the ER stress via the unfolded protein response (UPR), marked by upregulation of GRP78 and PERK-phosphorylation of eIF2α; ATF4 and CHOP are activated leading to ER-induced apoptosis. ER stress also leads to a release of calcium into the cytoplasm, which can directly activate caspase-12 and -4, as well as calpains, leading to apoptosis. High levels of calcium are also taken up by the mitochondria, depolarizing the membrane, resulting in apoptosis via cytochrome
C release. (
D) CDDP increases membrane fluidity and induces aggregation of components of the death inducing signalling complex (DISC) in the plasma membrane, resulting in extrinsic apoptotic activation. (
E) CDDP activates acid sphingomyelinase (aSMase) which leads to cytoskeletal collapse and production of ceramides, signalling cell death. Created with
BioRender.com (accessed on 11 November 2022).
The idea that CDDP accumulation may involve more than simple passive diffusion, however, arose through studies of CDDP resistance. In 2002, Ishida et al. found a connection between the high affinity copper uptake protein 1 (CTR1) and CDDP resistance. CTR1 knockouts in yeast resulted in a decreased CDDP uptake and provided resistance to the cells, allowing them to grow in otherwise toxic doses of CDDP [
35]. In addition, cells that were found to be resistant to copper transport were often found to be cross-resistant to CDDP, suggesting copper transporter involvement in CDDP uptake (
Figure 2). Indeed, yeast with CTR1 knockouts were found to have a 30% decrease in CDDP adduct formation compared to wild-type cells following CDDP treatment [
35]. Likewise, this phenomenon was mirrored in mammalian cell lines. Homozygous CTR1-knockout mouse ovarian cells were 8-fold more resistant to CDDP than wild-type cells, while heterozygous mutants displayed a 4-fold higher resistance [
35]. Katano et al. similarly demonstrated cross-resistance to CDDP in copper transport resistant cell lines [
36]. They found that CDDP-resistant cell lines had lower overall intracellular copper concentrations, and that these cells also showed lower rates of copper and CDDP accumulation [
36]. Although these studies demonstrate a correlation between CTR1 activity and CDDP uptake, it is still debated whether increased CDDP uptake by CTR1 translates into increased toxicity. In fact, some argue that while CTR1 may transport CDDP, it does not significantly contribute to CDDP toxicity. Holzer et al., for example, found that increased CTR1 expression increased CDDP uptake in ovarian cancer cells, but that there was no resulting increase in cytotoxicity or DNA adduct formation [
37]. Rather, they suggest that CTR1 mediated CDDP transport results in sequestration of CDDP in such a way that it cannot access the DNA, preventing CDDP transported by CTR1 from contributing to cytotoxicity [
37]. Holzer et al. subsequently demonstrated that treatment with CDDP actually leads to degradation of both exogenous and endogenous CTR1 on the plasma membrane, a phenomenon that was also observed upon copper treatment [
38,
39]. However, the internalization and degradation of CTR1 following CDDP treatment have been disputed by other groups [
33,
40,
41]. It is even argued that CTR1 does not act at all as a CDDP transporter. Kalayda et al. and Beretta et al. found that increased expression of CTR1 did not increase CDDP influx in ovarian carcinoma cells and cervical carcinoma cells, respectively, in direct opposition to previous work in support of CDDP uptake by CTR1 [
40,
42]. Another important point to consider is that CTR1 mediated transport of CDDP seems to be tissue specific [
33,
43]. For example, out of five small-cell lung cancer cell lines, only one showed a correlation between decreased CTR1 levels and increased resistance to CDDP [
44].
Another transporter seemingly implicated in CDDP accumulation is a Na
+/K
+-ATPase. In 1991, Andrews et al. found that CDDP uptake in OV2008 human ovarian carcinoma cells was decreased by 50% upon treatment with the Na
+/K
+-ATPase inhibitor ouabain [
45]. In addition, they found that depleting ATP reduced CDDP accumulation [
45]. This result has since been replicated by other groups. In 2006, Kishimoto et al. compared a parental CDDP-sensitive H4-II-E cell line with a CDDP-resistant H4-II-E cell line. They found through Western blotting that the CDDP-resistant cell line expressed lower levels of the Na
+/K
+-ATPase alpha subunit [
46]. In addition, upon depleting ATP using antimycin A, CDDP accumulation was decreased in the sensitive cells, but not in the resistant cells [
46]. This evidence suggests a potential role for active transport in CDDP accumulation that may be lost as a mechanism of CDDP resistance [
46]. Interestingly, Andrews et al. noticed in their study that the net Na
+/K
+-ATPase levels and activity were virtually identical in CDDP-sensitive and -resistant cells [
45]. They also found that CDDP accumulation was partially sodium dependent, and that it may depend on the electrochemical gradient maintained by Na
+/K
+-ATPase, in addition to the active CDDP transport [
45]. Incubation in low sodium media or other conditions that disrupted the sodium gradient resulted in a significant decrease in CDDP accumulation [
45]. Likewise, excess potassium resulted in a drastic 5-fold increase in CDDP influx, suggesting a relationship between the membrane polarization state and CDDP accumulation, in which membrane depolarization increases CDDP uptake [
47]. This relationship was further examined by Andrews et al. who showed that two CDDP-resistant cell lines had elevated basal membrane potentials in comparison to CDDP-sensitive cell lines [
47]. In summary, CDDP transport is likely a combination of passive, facilitated, and active transport mechanisms. However, it is unknown how much of a role each mechanism plays in overall CDDP transport. Further work must be carried out to reconcile the conflicting evidence presented in this review, as CDDP transporters could present as an attractive target for CDDP-resistant cancer cases.
3. DNA as the Primary Target of Cisplatin
The most well-studied mechanism of CDDP-mediated cell death is nuclear DNA damage following intracellular uptake. In the plasma, CDDP is relatively unreactive as the high chloride concentration (~100 mM) prevents the displacement of the chloride leaving groups [
48]. The intracellular chloride concentration, however, is much lower, varying between 5 and 60 mM. The decreased chloride concentration within the cell favours the aquation of CDDP, resulting in a di-aquo species [
49,
50,
51]. The resulting compound, (Pt(H
2O)
2(NH
3)
2)
2+, is a powerful electrophile that can bind a variety of cellular constituents, one of which being the highly nucleophilic N7-sites on the purine bases of the DNA [
50]. CDDP can fit well into the major groove of the DNA helix, allowing easy access for adduct formation [
50]. The major adducts formed by CDDP can be grouped into three major types: intrastrand crosslinks, interstrand crosslinks, and mono-functional adducts [
50] (
Figure 2A). The majority (~90%) of CDDP adducts are intrastrand crosslinks; 65% are 1,2-d(GpG) intrastrand crosslinks, 25% are 1,2-d(ApG) intrastrand crosslinks, and 5% are 1,3-d(GpXpG) intrastrand crosslinks. The remaining 5% of adducts are comprised of 2–5% interstrand d(GpG) crosslinks, and less than 1% are CDDP mono-functional adducts [
50,
52,
53,
54]. As the majority, intrastrand crosslinks are commonly thought to be the primary lesion responsible for CDDP cytotoxicity; however, the roles of interstrand crosslinks and mono-functional adducts have not been fully elucidated, and therefore, cannot be discounted when considering CDDP-mediated cell death [
9]. Indeed, recent studies provide evidence for interstrand crosslinks as significant contributors to CDDP toxicity and will be discussed later.
The ability of CDDP to inflict DNA damage has been known since the 1970s and is considered the principal mechanism of CDDP-mediated toxicity [
23,
55,
56,
57,
58]. Yet, the exact mechanisms of how CDDP DNA damage results in cell death remain unclear, in part due to the complex and multifaceted nature of how CDDP causes damage to the cell, and how the cell chooses to respond in turn. One long contested subject is the importance of replication and transcription stalling in CDDP-mediated cell death. Initially, it was thought that the main mechanism of cell death was inhibition of DNA synthesis, achieved through blocking DNA replication machinery by CDDP-DNA adducts [
59,
60]. In the late 1980s, studies by Sorenson and Eastman shifted this established paradigm away from inhibition of DNA synthesis towards prevention of RNA transcription as the primary culprit of CDDP related cell death [
61,
62,
63]. The results of these studies made two important points that casted doubt on the inhibition of DNA synthesis as a mechanism of CDDP-mediated cell death. First, cells treated with CDDP typically arrest within the G2 phase of the cell cycle, not the S phase as would be expected with inhibition of DNA synthesis [
61]. Second, in studies of DNA repair-deficient mouse ovarian cell lines, treatment with doses of CDDP known not to inhibit DNA synthesis were still found to be capable of inducing G2 growth arrest and apoptosis [
62]. In addition, they found that DNA was able to double even in cells that did not divide [
61,
63]. Rather, Sorenson and Eastman point to inhibition of transcription as the key mechanism of CDDP toxicity, suggesting that G2 arrest is a result of decreased whole-cell RNA and protein levels [
61,
63].
4. Regulation of Transcription by Cisplatin
The ability of CDDP to inhibit transcription by RNA polymerase II (Pol II) has been widely documented [
64,
65,
66,
67,
68,
69,
70]. Corda et al. provided early evidence of this phenomenon in the 1990s. They showed that, in vitro, transcription of a template strand was blocked by CDDP adducts, while the complementary strand was able to be transcribed [
67]. Later, they determined that ribonucleotide addition was differentially affected by different CDDP-DNA adducts. For example, d(GpG) intrastrand crosslinks inhibited Pol II to a higher degree than d(ApG) crosslinks [
68]. It was determined that d(GpG) lowered the binding affinity of Pol II to the DNA template, while no such effect was observed with d(ApG) crosslinks [
68]. Comparing interstrand and intrastrand crosslinks revealed that both types of CDDP adducts where capable of diminishing single nucleotide addition by pol II, and that both were able to irreversibly blocking nucleotide elongation [
69]. Interestingly, both the cis- and trans- isomers of DDP can inhibit DNA replication, decreasing DNA synthesis to a similar degree. Yet, the trans isomer is significantly less potent and dangerous to cancer cells than CDDP [
70]. This provides further evidence that inhibition of DNA replication is not the main culprit in CDDP-mediated cell death. Indeed, in 1995, Mello et al. provided in vivo evidence that CDDP blocked transcript elongation of a plasmid template in mammalian cells 2-fold more than transplatin [
70]. Furthermore, they provided evidence that the difference between CDDP and transplatin was not due to preferential repair of transplatin lesions by nucleotide excision repair (NER) or due to a decreased number of transplatin adducts compared to CDDP [
70]. Finally, 60–76% of transplatin adducts could be bypassed by Pol II, while only 0–17% of CDDP adducts could be bypassed by the enzyme [
70].
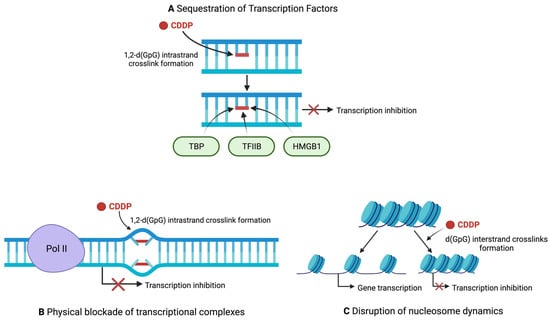
CDDP damage decreases transcription levels through three mechanisms: sequestration of transcription factors, physical blockade of transcriptional complexes, and through chromatin alterations [
71] (
Figure 3). In 1997, Vichi et al. were able to prove, through in vitro experiments utilizing reconstituted transcriptional systems, that the presence of CDDP-damaged DNA resulted in a 4-fold reduction in transcription compared to an undamaged template [
72]. Indeed, this finding was again validated by another group in 1999, providing further evidence that transcription can be repressed by CDDP-DNA adducts [
73]. Moreover, the TATA binding protein (TBP) was shown to directly bind CDDP-DNA adducts in nitrocellulose filter binding assays, and incubating CDDP-damaged DNA with TBP in a recombinant transcriptional system drastically reduced transcription of the undamaged template [
72]. Even when the template DNA contained TATA box motifs, TBP still preferentially bound CDDP-DNA adducts and only by re-introducing excess TBP into the assay reaction was the transcriptional repression alleviated [
72]. This suggests that CDDP-damaged DNA acts as a competitive inhibitor of the TBP–TATA box interaction and interferes with transcription through sequestering essential transcriptional machinery [
72] (
Figure 3A). This phenomenon was also seen with other components involved in the transcription complex such as TFIIB and TFIIH, however to a somewhat lesser degree than with TBP [
72]. Sequestration by CDDP-damaged DNA has since been demonstrated with other transcription factors. For example, the rRNA transcription factor known as the upstream binding factor (UBF) is also sequestered by CDDP-DNA damage [
74,
75]. UBF tightly binds 1,2–dGpG intrastrand crosslinks, preventing rRNA transcription in vitro [
74,
75]. In this way, sequestering UBF could halt the production of rRNA, a necessary process during cell proliferation and growth, and contribute to CDDP toxicity [
75]. In addition, the Y-box, high mobility group box 1 (HMGB1), and the structure specific recognition protein 1 (SSRP1) in complex with Spt16 are all important transcription factors that have each been shown to specifically bind to CDDP-DNA adducts with high specificity, and likely contribute to transcription inhibition by CDDP [
76,
77,
78] (
Figure 3A).
Figure 3. Mechanisms of transcription inhibition by CDDP. (
A) CDDP hinders transcription by sequestering transcription factors away from promoter regions. (
B) Bulky CDDP lesions act as a physical roadblock to transcription machinery, preventing transcript elongation. (
C) CDDP interstrand crosslinks hinders nucleosome mobility, preventing chromatin remodelling and maintaining condensed chromatin that prevents transcription. Red crosses represent inhibition of transcription. Created with
BioRender.com (Accessed on 11 November 2022).
Initial evidence for the physical blocking of transcription machinery directly by CDDP adducts was provided by Mymyrk et al. in 1995, when they showed that CDDP adducts could block NF1 binding to template DNA [
79]. Indeed, in vitro studies confirmed that CDDP-damaged DNA could block NF1 transcription factor binding. The template DNA utilized had no chromatin structure, confirming that CDDP was not inducing a change in chromatin conformation that blocked NF1 binding in this experiment [
79]. In other words, CDDP-adducts alone were shown to be sufficient to directly block transcription factor binding, either through steric hindrance or through DNA helix distortion [
79]. A clearer picture of how CDDP blocks Pol II came from a structural study of yeast Pol II in complex with a CDDP-damaged nucleotide scaffold [
80]. In this study, a d(GpG) intrastrand lesion was specifically placed on the +2/+3 position of the template scaffold, with the +1 position being where ribonucleotide addition takes place. The authors found that the bulky CDDP lesion prevent the afflicted base from being stably accommodated in the Pol II active site. Indeed, backtracking of the polymerase occurred when the lesion was attempted to be placed in the active site [
80]. The authors therefore proposed that CDDP acts as a translocation barrier of sorts, preventing the adducted base from correctly binding the active site of Pol II, thereby physically stopping Pol II in its tracks [
80] (
Figure 3B). Another observation was the misincorporation of AMP by the Pol II elongation complex at the site of lesion, creating a G-A mismatch [
80]. The phenomenon of AMP misincorporation by stalled polymerases is well documented, and is known as the A-rule [
81]. However, Pol II stalling was shown to occur regardless of the presence of the G-A mismatch, further suggesting a bulky lesion translocation barrier as the main feature in Pol II stalling [
80].
The final mechanism thought to contribute to transcriptional inhibition is the disruption of nucleosome dynamics. The condensation of eukaryotic DNA into chromatin provides a dual function, allowing the DNA to fit within the nucleus of the cell acting as a level of regulation for gene expression [
82]. The nucleosome is the base unit of chromatin structure and consists of DNA coiled around histone core proteins. The wrapped DNA becomes inaccessible to the transcription machinery, and thus, chromatin remodelling through nucleosome mobility is critical for ensuring successful gene transcription [
83]. CDDP has been shown to interfere with nucleosome dynamics and chromatin remodelling both in vitro and in vivo (
Figure 3C). As early as 1995, observations were made that CDDP treatment in mouse mammary cells prevented both nucleosome shifting and transcription itself; however, a mechanistic explanation of how CDDP prevented nucleosome mobility was not given [
79]. More recently, structural studies have been able to provide some insight into the mechanism of how CDDP locks nucleosomes. X-ray crystallographic studies by Ober and Lippard have shown that intrastrand crosslinks positioned inside the nucleosome adopt a position facing inwards, toward to the core [
84,
85]. They suggest that the sliding of nucleosomes along Pt-damaged DNA would bend the lesion into an unfavourable position [
86]. In this way, CDDP crosslinks can lock nucleosomes in place and prevent chromatin remodelling. Interestingly, 1,2 intrastrand crosslinks were found to have a stronger inhibitory effect on nucleosome mobility than 1,3 crosslinks, as the 1,2 crosslinks bend at a more severe angle in the nucleosome [
86]. Yet, despite being able to reduce nucleosome mobility, adducting nucleosome DNA with CDDP was unable to prevent transcription by bacterial RNA polymerases. This finding led Ober and Lippard to conclude that nucleosome dynamics must not be a mechanism of transcriptional inhibition by CDDP [
86]. Others, however, would disagree with this conclusion. In a 2021 study, Moon and colleagues found that CDDP locks nucleosomes in place and fastens chromatin. Even in conditions of high salt and mechanical force, which can release untreated nucleosomes, CDDP treatment resulted in nucleosome fixation, fully preventing the disassembly of the histones [
87]. This fixation also seemed to be permanent, remaining even after exposure to 3M NaCl [
87]. Important to note, however, is the concentration of CDDP used in this study. Treatments with 3.3 mM CDDP, and to a lesser extent 0.1 mM, were capable of locking nucleosomes and suppressing transcription of genes contained within the nucleosomes [
87]. The therapeutic range for CDDP is thought to lie between 1.0 and 5.0 mg/mL, or roughly between 3 and 16 µM [
88]. Concentrations of 3.3 mM and even 0.1 mM are therefore much too high to be physiologically relevant and must be considered when analyzing these findings. Another question to consider is how intrastrand and interstrand crosslinks differ in their ability to suppress nucleosome mobility. In a 2013 study by Zhu, Song and Lippard, nucleosome shifting was observed upon heat treatment. Nucleosomes pretreated with CDDP, however, had a drastically reduced ability to heat shift [
89]. Interestingly, nucleosomes with 1,2-d(GpG) intrastrand crosslinks were readily shifted upon heat exposure, while nucleosomes with interstrand crosslinks demonstrated a significantly reduced ability to shift [
89]. However, both types of crosslinks were capable of inhibiting transcription in vivo regardless of their ability to prevent nucleosome shifting [
89]. These results point to a potential mechanistic difference between CDDP intrastrand and interstrand crosslinks. Perhaps reduced nucleosome mobility is a mechanism used by interstrand, but not intrastrand, crosslinks to inhibit transcription. Alternatively, reduced nucleosome mobility may not play a significant role in the inhibition of transcription, acting because of CDDP-mediated damage rather than a cause. In either case, these results provide evidence that interstrand crosslinks may play a larger role in CDDP-mediated toxicity, a role that historically was attributed solely to intrastrand crosslinks.
How CDDP mediates cell death depends not only on the DNA lesions themselves but also on how the cell chooses to respond to the damage. In cases of short-spanned tolerable damage, the cell can activate cell cycle arrest and repair mechanisms. In cases of unmitigated sustained damage, the cell can activate pro-apoptotic pathways [
90]. DNA damage by CDDP is recognized by various sensor proteins, including but not limited to HMGB1, hMutSa, hUPB, and TBP [
91,
92,
93]. Transcriptional inhibition itself may also act as a pro-apoptotic damage signal, as it may increase the ratio of pro-apoptotic transcripts—which tend to have longer half-lives—to anti-apoptotic transcripts [
71]. The recognition of CDDP DNA damage by these sensors leads to the activation of both intrinsic and extrinsic apoptotic pathways. In brief, CDDP damage results in action of the ATR kinase, which phosphorylates serine-15 on p53, resulting in its activation [
94]. P53 can also be activated via ERK, which itself is activated by MAPK upon CDDP damage. Ultimately, activation of p53 results in upregulation and activation of pro-apoptotic proteins, which result in cytochrome
C release from the mitochondria and activation of caspase-9 [
95]. Caspase-9 will in turn activate the executioner caspases-3 and -7 leading to cell death [
96,
97]. CDDP may also activate a p53-independent form of intrinsic apoptosis that stems from the activation of the c-abl tyrosine kinase, resulting in the activation of p73 [
98]. P73, a p53-like protein, activates intrinsic apoptosis similarly to p53, through cytochrome
C release and activation of executioner caspases [
98]. Finally, CDDP induction of MAPK also triggers extrinsic apoptosis through the JNK and p38 pathways, which culminate in Fas ligand (FasL) gene expression [
99,
100]. FasL in turn binds the Fas receptor (FasR) on the plasma membrane, resulting in the formation of the death-inducing signalling complex (DISC) followed by apoptosis [
99,
100].
5. Non-Nuclear Targets of Cisplatin
While DNA damage is historically considered to be the primary mechanism of CDDP-induced cell death, there is evidence to suggest that CDDP may induce apoptosis independently of nuclear DNA (nDNA) damage. For instance, enucleated HNSCC cell cytoplasts remain as sensitive to CDDP as their nucleated parental cell line [
101]. In addition, it has been reported that CDDP can lead to caspase-3 activation even in enucleated cytoplasts, suggesting possible apoptotic initiation in the cytoplasm rather than in the nucleus [
102,
103]. In fact, in testicular germ cell tumours the DNA damage response proteins ATM, ATR, and DNA-PK are not required for initiation of apoptosis in response to CDDP treatment [
104]. Thus, it is necessary when evaluating CDDP-mediated cell death to look for other cellular targets. One target that plays a large role in CDDP toxicity is mitochondrial function (
Figure 2B). Interestingly, CDDP has a higher propensity for forming mitochondrial DNA (mtDNA) adducts than nDNA adducts, with mtDNA lesions being present in levels 300–500 fold higher than nDNA lesions [
101]. Furthermore, in Chinese hamster ovary cells, there is not only higher initial binding of CDDP to mtDNA than nDNA, but also a lower rate of excision of mtDNA adducts than nDNA adducts, resulting in higher levels of mtDNA damage compared to nDNA [
105]. Likewise, cells that have been depleted of their mtDNA are 4–5-fold more resistant to CDDP than their parental cell lines [
101,
106].
Mitochondrial damage via CDDP adducts on mtDNA results in oxidative stress and the production of reactive oxygen species (ROS) [
107,
108,
109,
110]. The production of ROS by CDDP has shown to be majorly dependent on mitochondrial damage (
Figure 2B). Cells with depleted mtDNA do not generate nearly as high levels of ROS upon exposure to CDDP, and ROS generated following CDDP treatment is localized to the mitochondria [
110,
111]. In addition, pre-treatment of cancer cell lines with N-acetyl cysteine (NAC), an antioxidant, reduces sensitivity to CDDP alleviating its cytotoxic effects. CDDP adducts on mtDNA block mtDNA transcription, leading to a reduction in the translation of proteins necessary for the electron transport chain (ETC) [
112]. Impaired ETC function can in turn generate high levels of mitochondrial ROS, which can damage a variety of cellular constituents including mtDNA, proteins, and lipids. ROS can further impair the mitochondria and the ETC, causing a positive feedback loop generating even more ROS and culminating in apoptosis via the intrinsic or extrinsic pathways [
110,
113,
114]. For example, ROS can increase mitochondrial membrane permeability, disrupt mitochondrial membrane potential, and facilitate cytochrome
C release from the mitochondria through activation of Bak and Bax [
113,
114,
115,
116,
117]. ROS can also trigger the extrinsic apoptotic pathway by activating the death receptors TRAIL-R1/2, FasR, and TNF-R1 on the plasma membrane [
113].
A 2019 study by Kleih and colleagues gave further insight into the mechanisms of CDDP-mediated mitochondrial ROS generation. They determined, in ovarian cancer cell lines, that CDDP-sensitive cells tended to have higher baseline mitochondrial content and mitochondrial ROS than CDDP-resistant cells [
118]. Moreover, they found that treatment with CDDP actually stimulates mitochondrial biogenesis and suggest that CDDP treatment triggers an increase in mitochondrial content and subsequent increase in mitochondrial ROS production [
118]. Indeed, preventing mitochondrial biogenesis through knockdown of PGC1a or TFAM, important factors for mitochondrial biogenesis, resulted in reduced susceptibility to CDDP [
118]. This is paralleled by the fact that higher mitochondrial content, represented by high TFAM expression, correlates with a better 5-year survival in ovarian cancer patients [
118]. However, this study was conducted only in ovarian cancer and should be repeated in other cancer types. In addition, it is unclear whether ROS production from CDDP treatment is a result of solely increased mitochondrial content, through damaged ETC, or a combination of both [
118].
The endoplasmic reticulum (ER), an arm of the secretory pathway involved in protein folding and quality control, lipid synthesis, and calcium storage/homeostasis represents a cytoplasmic target of CDDP [
119,
120] (
Figure 2C). As part of its role in maintaining protein quality control, the ER is equipped with mechanisms to overcome disturbances to protein homeostasis. One such mechanism is the unfolded protein response pathway that becomes activated in response to ER stress [
121]. Under normal conditions, the ER chaperone GRP78 acts to facilitate proper protein folding, as well as negatively regulate UPR stress sensors [
122]. However, upon ER stress, GRP78 becomes activated, in turn activating transcription factor 6 (ATF6), inositol-requiring protein 1α (IRE1α), and protein kinase RNA-like endoplasmic reticulum kinase (PERK) [
122]. These stress sensors initiate responses that promote maintenance of protein homeostasis and cell survival, or in cases of extreme unmitigated ER stress, apoptosis [
121,
123]. For example, PERK acts to reduce global protein translation by phosphorylating eukaryotic translation factor 2α (p-eIF2α), while IRE1α initiates selective degradation of certain mRNAs. Both of these sensors work to reduce the total amount of protein moving through the ER, allowing the ER to remediate the imbalance of unfolded proteins and promote cell survival [
123,
124]. In cases of chronic ER stress, ATF4, a downstream factor of the PERK pathway, upregulates the expression of C/EBP homologous protein (CHOP). CHOP promotes apoptosis through inducing expression of pro-apoptotic factors while simultaneously downregulating anti-apoptotic factors [
124,
125]. In addition, IRE1α activation results in the activation of caspase-12, an ER-specific caspase that can induce apoptosis upon prolonged ER stress via activation of caspases-9 and -3 [
126,
127]. ATF6, upon ER stress, translocates to the Golgi apparatus, where it is subsequently cleaved and transported to the nucleus. In the nucleus, the cleaved portion of ATF6 induces expression of different UPR effectors, including ER chaperones and CHOP [
128]. CDDP has been shown to induce aggregation of misfolded proteins within the ER, pushing the cell towards ER stress [
129] (
Figure 2C). Likewise, CDDP upregulates the expression of UPR markers, such as GRP78, caspase-12, GADD34, and CHOP [
102,
129,
130]. Interestingly, this effect has been observed both in various cancer cells lines and in enucleated cytoplasts, confirming again that CDDP can trigger the cytoplasmic apoptotic pathway independent of nuclear damage [
102]. Similarly, inhibition of caspase-12 activity has been found to reduce the percentage of apoptotic cells upon treatment with CDDP compared to controls, further implicating ER stress and caspase-12 activity as a notable mechanism of CDDP-mediated cell death [
131].
ER stress induced by CDDP also contributes to cytotoxicity through the efflux of stored calcium from the ER into the cytoplasm, drastically increasing its cytoplasmic concentration [
132]. Increased calcium in the cytoplasm leads to activation of calpains, calcium dependent cysteine proteases, as well as caspase-4 [
133] (
Figure 2C). Calpains aid in triggering cytochrome
C release through the activation of the pro-apoptotic factor Bid, and are also thought to play a role in caspase-4 activation, both of which ultimately result in apoptosis [
134]. Indeed, blocking calcium release from the ER through inositol triphosphate receptor inhibitors proved to prevent caspase-4 and calpain activation, while simultaneously mitigating apoptotic activation by CDDP [
132]. Furthermore, increased calcium in the cytoplasm can cause calcium uptake into the mitochondria through various transporters [
135]. Inside the mitochondria, the increased calcium concentration leads to the generation of ROS, which further damages the mitochondria and activate apoptosis [
135,
136].
Despite previous evidence, much remains to be understood regarding the role of ER stress in CDDP cytotoxicity, as certain studies have shown that ER stress can play a protective role against CDDP, rather than a cytotoxic one [
120,
130,
137]. In a 2017 study, the HIV protease inhibitor saquinavir, known to induce ER stress in ovarian cancer cells, increased the IC
50 of CDDP when used in combination [
138]. Because of inducing ER stress, saquinavir also upregulated the expression of genes involved in autophagy, a common response to ER stress [
138,
139]. The authors suggest that increased autophagy resulting from ER stress assists in protecting the cell from CDDP by aiding in clearance of damaged cellular constituents [
138]. Taken together, it is likely that ER stress affects the action of CDDP depending on the degree of ER stress. In low levels, ER stress seems to play a protective role, ameliorating the toxic effects of CDDP by inducing autophagy and other pro-survival pathways. In cases of consistent, high-level ER stress, however, cell death mechanisms in response to CDDP are the favoured response. Further investigation should be done on the interplay between ER stress and CDDP activity, as the ER stress response could provide a promising target for cancers with low sensitivity to CDDP.
Other cytoplasmic targets of CDDP are cellular lipids, particularly those of the plasma membrane. CDDP has been shown to interact with phospholipids and phosphatidylserine both in liposome membrane models and in human erythrocytes [
140,
141]. In fact, human erythrocytes change shape as a response to treatment with CDDP, indicating that CDDP may affect plasma membrane structure [
142]. Yet, despite the interaction of CDDP with the erythrocyte membrane, and subsequent change in membrane integrity, there was no change in the fluidity of the membrane [
142]. However, other studies have demonstrated changes in membrane fluidity because of CDDP treatment. Studies utilizing liposomes reveal, via atomic force spectroscopy, that CDDP induced a reduction in membrane fluidity compared to untreated liposomes [
143]. In addition, lateral diffusion within the lipid bilayer has also been shown to decrease upon treatment with CDDP [
144]. In contrast, studies involving HT29 human colon cancer cells provide evidence that CDDP actually leads to an increase in membrane fluidity [
145,
146]. Indeed, treatment with CDDP resulted in a rapid increase in membrane fluidity due to CDDP-mediated activation of acid sphingomyelinase (aSMase) activity [
145,
146]. Additionally, CDDP induced the formation and activation of the death-inducing signalling complex (DISC) on the plasma membrane. CDDP has been shown to cause aggregation of the Fas death receptor (FasR), a component of the DISC, in the plasma membrane [
145]. Furthermore, CDDP causes accumulation and re-localization of pro-caspase-8 and FADD, other DISC components, along with the FasR, to lipid rafts. In this way, CDDP is likely inducing ligand independent activation of DISC, resulting in capase-8 activation and apoptotic signalling [
145] (
Figure 2D). Indeed, treatment with nystatin, a compound that sequesters cholesterol, prevented the redistribution of FasR, FADD, and pro-caspase-8 to lipid rafts upon CDDP treatment, and reduced CDDP-mediated cell death as a result [
145]. The increase in membrane fluidity resulting from CDDP treatment is thought to aid in the redistribution of DISC components into the lipid rafts [
145]. In fact, pre-treatment with cholesterol, which decreases membrane fluidity, resulted in the reduced re-localization of DISC components to lipid rafts and reduced apoptosis even in the presence of CDDP, further demonstrating the importance of membrane fluidity in CDDP-mediated DISC activation [
145]. Interestingly, analysis of the plasma membrane composition of CDDP-resistant and CDDP-sensitive cell lines reveal that the CDDP-resistant cells had higher levels of phosphatidyl choline and lower levels of cholesterol in their plasma membranes, decreasing the fluidity [
147]. The decrease in membrane fluidity in CDDP-resistant cells may act as a compensatory resistance mechanism to protect against the increase in membrane fluidity caused by CDDP [
147]. Finally, CDDP induction of aSMase activity results not only in increased membrane fluidity but also in the production of ceramides [
145,
146] (
Figure 2E). Ceramides are cell death signalling lipids shown to be involved with Bax accumulation in the mitochondria and permeabilization of the outer mitochondrial membrane and activation of caspase-3, ultimately resulting in apoptosis [
148]. In this way, production of ceramides by CDDP-mediated activation of aSMase may contribute to cytotoxicity by CDDP.
In addition to the plasma membrane, CDDP also induces damage to the cytoskeleton. Cancer metastasis demands efficient remodelling of the cytoskeletal components, microfilaments, intermediate filaments, and microtubules, in order to allow for cell migration and invasion [
149]. CDDP has been shown to directly interfere with the cytoskeleton network, causing its collapse and subsequent aggregation in the cytoplasm [
150] (
Figure 2E). For example, CDDP causes vast remodelling of the actin microfilament network, causing loss of filopodia and an increase in cortical stress fibers [
151]. In addition, CDDP causes the destabilization of actin filaments anchored to the plasma membrane, causing its re-localization to the cytoplasm. This effect is mediated through ezrin, an actin binding protein with roles in anchoring actin to the plasma membrane, that becomes deactivated by CDDP via CDDP-induced aSMase activity [
151]. Indeed, knockdown of aSMase activity abrogates the actin and ezrin re-localization induced by CDDP. In addition, treatment with ceramides alone cause the same effects of ezrin inactivation as by CDDP, showing that ceramides produced by CDDP-activated aSMase are responsible for the disruption of the actin network [
151].
CDDP also displays activity against microtubules. Through direct binding to tubulin, CDDP promotes tubule depolymerization [
152]. Furthermore, CDDP prevents assembly of tubulin into microtubules, which causes accumulation of tubulin aggregates unable to participate in cellular functions such as migration and mitosis [
153]. In fact, a study investigating the role of CDDP in cytoskeletal function showed that CDDP treatment increased cell stiffness, and decreased cell migration, invasion, and colony formation as a result of its cytoskeletal effects [
154]. In this way, CDDP exerts anti-tumour activity by disrupting cytoskeletal remodelling.
6. Limitations of the Use of Cisplatin
The introduction of CDDP into treatment regimens revolutionized cancer therapy. However, CDDP has significant drawbacks that limit its usefulness as an anti-cancer agent. The most significant limitations of CDDP are its severe side effects, which manifest primarily through nephrotoxicity, ototoxicity, and neurotoxicity (
Table 1). Among them, nephrotoxicity is considered the most significant dose-limiting factor for discontinuance of CDDP with roughly one-third of all patients developing acute kidney injury in response to CDDP treatment [
155,
156,
157,
158]. It has been shown that the kidneys accumulate higher levels of CDDP than any other tissues, and that CDDP action on the kidneys results in reduced glomerular filtration rate and increased serum creatinine levels, indicating impaired renal function [
158,
159]. Through the same mechanisms described above, CDDP triggers an apoptotic response in the tubular cells of the proximal and distal tubules of the nephron [
160,
161]. In addition, CDDP treatment results in ROS production that further contributes to the induction of apoptosis via activation of mitochondrial damage and the extrinsic apoptotic pathway [
162]. Furthermore, CDDP has been shown to induce an acute inflammatory response in renal tubular cells. CDDP treatment releases pro-inflammatory mediators, with TNFα considered the major factor involved in CDDP-induced inflammation [
163,
164]. Additionally, TNFα initiates pro-apoptotic signalling through its interaction with TNFR2 [
164,
165]. Finally, CDDP significantly damages the renal vasculature. Through ischemic injury and induced vasoconstriction, CDDP decreases the glomerular filtration rate and brings about hypoxic injury [
166,
167].
Another dose-limiting side effect of CDDP is the resulting ototoxicity [
168]. Anywhere from 20% to 70% of patients experience CDDP-related ototoxicity, with this effect being more prominent in children [
168,
169]. CDDP induces ototoxicity through induction of ROS and apoptotic cell death within the hair cells of the ear [
170,
171]. As a result, 40–80% of patients develop permanent hearing loss [
172]. Interestingly, the OCT2 transporter has been reported to be involved in mediating both CDDP nephrotoxicity and ototoxicity, by transporting CDDP into both renal tubule cells and hair cells of the inner ear [
173,
174]. In fact, blocking OCT2 activity results in markedly reduced cellular uptake of CDDP by these cells in vitro, and reduced nephro- and ototoxicity in vivo [
173,
174]. OCT2, while expressed in these cells, is not typically expressed to a significant degree in most tumour types that CDDP is used to treat [
173]. Therefore, OCT2 may prove to be a useful target for reducing off target toxicity without diminishing CDDPs cytotoxic effects against tumour cells.
Neurotoxicity is an additional side effect of CDDP with severe consequences for patients. CDDP has been shown to accumulate and cause damage to the dorsal root ganglia causing morphological changes and resulting in peripheral neuropathy [
175]. Sensory neurons are more significantly affected by CDDP, with common symptoms being paresthesia and dysesthesia in the extremities [
175,
176]. Neuropathic symptoms can continue to worsen up to 6 months post-completion of CDDP treatment and recovery tends to occur very slowly, with some patients reporting neuropathy symptoms up to 20 years post-treatment [
177,
178,
179].
Finally, hepatotoxicity and cardiotoxicity have also been observed upon CDDP treatment; however, these are considered rare [
180,
181] (
Table 1). More general side effects include nausea and vomiting, diarrhea, fever, weight loss, and transient hair loss, among others [
182,
183].
Another major limitation of CDDP is acquired resistance following repeated exposure, which is facilitated by a variety of mechanisms [
184]. One way in which cells become resistant to CDDP is through decreased CDDP accumulation [
184]. Many studies have shown that CTR1, the copper transport protein involved in CDDP import, plays an important role in CDDP resistance. CDDP-resistant cell lines have shown to have decreased expression of CTR1, which results in decreased intracellular accumulation of CDDP [
40,
185]. CTR2 may also play a role in CDDP resistance by stimulating the cleavage of CTR1 by cathepsin L/B and further reducing accumulation of CDDP [
186].
Increased CDDP efflux is another mechanism of acquired resistance [
184]. CDDP efflux is mediated by the copper efflux transport proteins ATP7A and ATP7B (
Figure 2). Elevated levels of ATP7A/B correlate with increased resistance to CDDP in vitro and with poor prognosis in patients [
187,
188,
189]. In addition, knocking out ATP7A increases the efficacy of CDDP in vitro [
190,
191]. Finally, multidrug resistance-associated proteins, such as MRP2, also contribute to enhanced CDDP efflux (
Figure 2). MRP2 works to export CDDP bound to the cellular antioxidant glutathione (GSH), and increased MRP2 expression contributes to CDDP resistance [
192,
193]. Similarly, increased drug detoxification through GSH activity also contributes to CDDP resistance [
194]. GSH binds activated CDDP in order to deactivate and facilitate its removal via MRP exporters and other detoxifying enzymes [
194]. GSH also provides resistance to CDDP independently of detoxification through its activity as a ROS scavenger [
195]. By reducing the high levels of ROS induced by CDDP, GSH ameliorates the cytotoxic oxidative stress [
195,
196]. Cells resistant to CDDP have shown to have increased expression of GSH and additional enzymes involved in CDDP detoxification and ROS scavenging [
197]. In fact, reducing intracellular GSH re-sensitizes resistant cells to CDDP [
198]. In addition, increased levels of NF-E2-related factor 2 (Nrf2), a transcription factor involved in the regulation of GSH and MRP, are associated with increased CDDP resistance in vitro and in vivo [
199,
200,
201]. Nrf2 also contributes to CDDP resistance through its interaction with the ubiquitin binding protein p62 [
202]. P62 activation by Nrf2 upregulates autophagy and clearing of misfolded proteins via the proteasome, reducing ER stress and further increasing resistance to CDDP [
203,
204].
Increased DNA damage repair is another mechanism of CDDP resistance [
205]. Intrastrand crosslinks are primarily repaired through the NER pathway [
206,
207]. Crosslink formation distorts the shape of the DNA helix, allowing them to be detected by a complex of xeroderma pigmentosum C (XPC) and RAD23B [
208,
209,
210]. Upon detection of the lesion, XPC-RAD23B recruits transcription factor II H (TFIIH), a complex of multifunctional protein subunits [
211]. The XPB subunit unwinds the helix, while XPD locates the lesion and, upon stalling at the site of the crosslink, signals for the recruitment of the pre-incision complex, consisting of RPA, XPA, and XPG [
212]. ERCC1-XPF, a 5′-endonuclease, is then recruited to the protein scaffold by interaction with XPA, where it cuts the DNA strand 5′ to the lesion [
213]. A 3′ incision is then made by XPG, and together this process is known as dual excision [
214]. Following excision, the lesion is released with TFIIH and repair is completed by a series of DNA polymerases (δ, κ, or ε.) [
215,
216]. Finally, the nick in the helix is repaired most commonly by DNA ligase I [
206]. It has been shown that increased expression of NER-related genes is associated with increased CDDP resistance. This effect has been observed with a multitude of NER genes, such as XPF, XPA, and XPG; however, the strongest correlation is seen with ERCC1 [
217].
Homologous recombination (HR) is another DNA repair pathway involved in CDDP lesion repair. The pathway for repair of CDDP interstrand crosslinks involves the formation of double strand breaks (DSB) in the DNA [
205]. These DSBs are then in turn repaired by specialized DSB-repair pathways such as HR [
205]. BRCA-1 and -2 are essential to HR function and promote DNA end resection and recruitment of other HR components [
218]. Defects in these genes are relatively common in certain cancer types, and increase the sensitivity of tumour cells to CDDP [
219]. However, it has been shown that, in ovarian cancer cells, HR can be rescued by subsequent mutations that restore BRCA1/2 wild type activity, increasing resistance to CDDP as a result [
220,
221].
Finally, the mismatch repair (MMR) pathway, a DNA repair response that corrects single nucleotide mismatches, can also recognize CDDP lesions [
222,
223]. Interestingly, MMR cannot effectively repair CDDP adducts, as it causes replacement of the base opposite to the lesion rather than the damaged based itself [
224]. As a result, the original damage source is maintained, and the MMR pathway can start again. This process is described as a futile cycle of repair, and results in the generation of genotoxic DSBs and apoptotic signalling [
224,
225]. In this way, MMR actually contributes to CDDP cytotoxicity in tumour cells, and its downregulation results in acquired resistance, as defective MMR is associated with decreased CDDP cytotoxicity and poor prognosis in patients [
226,
227,
228].
This entry is adapted from the peer-reviewed paper 10.3390/ijms232315410