Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Hydrogen sulfide (H2S), which is a gasotransmitter, can be biosynthesized and participates in various physiological and biochemical processes in plants. H2S also positively affects plants’ adaptation to abiotic stresses.
- hydrogen sulfide
- resistance
- cysteine residues
- plant growth regulator
1. Biosynthesis Pathway of H2S
The biosynthesis of H2S includes both non-enzymatic and enzymatic pathways.
1.1. Non-Enzymatic Pathway
The non-enzymatic pathway of H2S biosynthesis occurs due to the reaction of thiols or thiol-containing compounds with other molecules [1][2][3][4]. Glutathione (GSH) reduces inorganic polysulfides or hydrolyzed inorganic sulfide salts, i.e., sodium sulfide (Na2S) or sodium hydrosulfide (NaHS) with water to produce H2S [2][3][4]. Cysteine is the preferred substrate for the non-enzymatic production of H2S, and the process is catalyzed by iron and vitamin B6 [5].
1.2. Enzymatic Pathway
The trans-sulfuration pathway is the primary source of endogenous H2S and is the only way to produce endogenous cysteine, involving cellular sulfur metabolism and redox regulation [6] (Figure 1). Methionine, acting as a substrate, is catalyzed to produce cysteine eventually, with H2S as the byproduct. Cystathionine-β-synthase (CBS, EC 4.2.1.22), cystathionine-γ-lyase (CSE, EC 4.4.1.1), cysteine aminotransferase (CAT, EC 2.6.1.3), and 3-mercaptopyruvate sulfurtransferase (3-MST, EC 2.8.1.2) are all involved in this pathway. CBS and CSE are only located in the cytoplasm, while CAT and 3-MST can be present in the cytoplasm and mitochondria [7]. In the first irreversible step of converting methionine to cysteine, CBS catalyzes the condensation of homocysteine with serine (Ser) or cysteine to form cystathionine. The main role of CSE in the trans-sulfuration pathway is in the conversion of cystathionine to cysteine and α-ketobutyrate. CBS can produce H2S through β-substitution reactions [7][8][9]. Similarly, CSE can produce H2S via the β-elimination reaction with cysteine or the γ-replacement reaction between two homocysteine molecules. There is also a mechanism for H2S production, mediated by CAT and 3-MST in the transsulfuration pathway. CAT catalyzes l-cysteine and α-ketoglutarate to form 3-mercaptopyruvate (3-MP) and glutamate. The sulfur group of 3-MP is then transferred to 3-MST-accepting nucleophilic Cys247 in the presence of 3-MST, to produce 3-MST-bound persulfide and pyruvate. After this stage, the MST-persulfide reacts with thiols or is reduced by thioredoxin (Trx) to form H2S [10][11]. In addition, 3-MST transfers the sulfur group from 3-MP to cyanide, to form thiocyanate [12]. Similar to the CAT/3-MST pathway, there is also a d-amino acid oxidase (DAO, EC 1.4.3.3)/3-MST pathway to generate H2S (Figure 1). DAO metabolizes d-cysteine into 3-MP, which is metabolized into H2S by 3-MST [13].
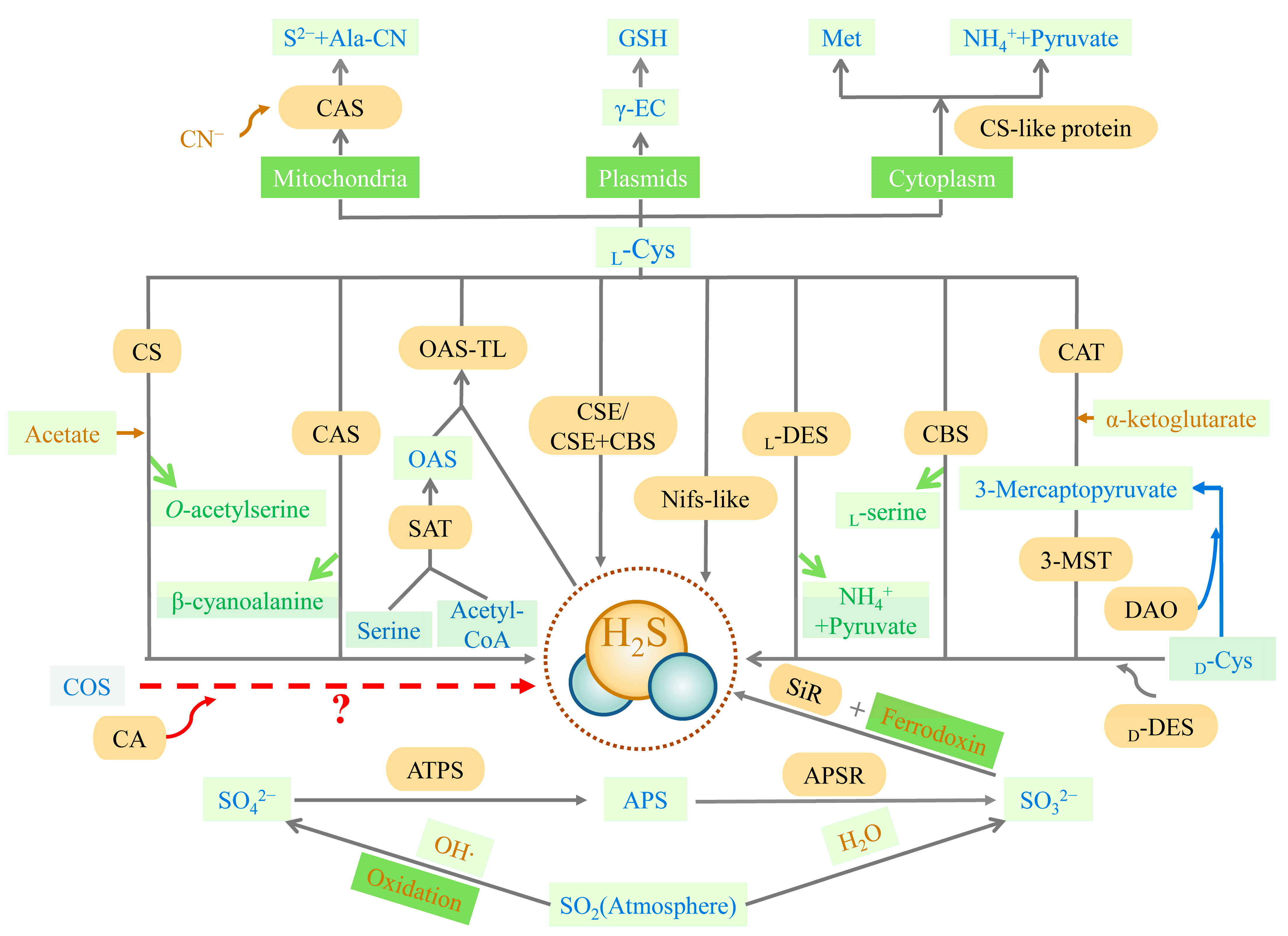
Figure 1. Synthesis of H2S. APS (adenosine 5′-phosphosulfate), APSR (APS reductase, EC 1.8.99.2), ATPS (ATP sulfurylase, EC 2.7.7.4), CA (carbonic anhydrase, EC 4.2.1.1), CAS (l-3-cyanoalanine synthase, EC 4.4.1.9), CAT (cysteine aminotransferase, EC 2.6.1.3), CBS (cystathionine-β-synthase, EC 4.2.1.22), COS (carbonyl sulfide), CN− (cyanide), CS (O-acetyl-l-serine via cysteine synthase, EC 4.2.99.8), CSC (hetero-oligomeric cysteine synthase complex), CSE (cystathionine-γ-lyase, EC 4.4.1.1), DAO (d-amino acid oxidase, EC 1.4.3.3), d-Cys (d-cysteine), d-DES (d-cysteine desulfhydrase, EC 4.4.1.15), γ-EC (γ-glutamylcysteine), GSH (glutathione), HO· (hydroxyl radicals), H2O (water), H2S (hydrogen sulfide), l-Cys (l-cysteine), l-DES (l-cysteine desulfhydrase, EC 4.4.1.1), Met (methionine), 3-MST (3-mercaptopyruvate sulfurtransferase, EC 2.8.1.2), NH4+ (ammonium), Nifs-like (nitrogenase Fe-S cluster-like), OAS (O-acetyl-l-serine), OSATL (O-acetylserine(thiol)lyase, EC 2.5.1.47), S2− (Sulfide), SAT (serine acetyltransferase, EC 2.2.1.30), SiR (sulfite reductase, EC 1.8.7.1), SO2 (sulfur dioxide), SO32− (sulfite), SO42− (sulfate). The closed lines filled with yellow represent enzymes. Rectangles represent substance/organelle reaction conditions. Rectangles filled with light green represent substances (wherein the blue-lettered cells are pivotal substances, yellow-lettered cells are involved in the synthesis of substrates, and green-lettered cells are reaction synthesis byproducts). Rectangles filled with dark green represent substance organelles/reaction conditions (wherein the white-lettered cells are organelles and the yellow-lettered cells are reaction conditions). Yellow arrows represent the substrates involved in the reaction, green arrows represent byproducts of the reaction, and red arrows represent the need for verification by further research. The reactions involved in the blue arrows are not currently found in plants.
In plants, the synthesis pathway of H2S can be divided into five types, according to the different substrates. These are cysteine degradation and sulfite reduction, cyanide detoxification, iron-sulfur cluster turnover, and carbonyl sulfide (COS) conversion [14] (Figure 1). Similar to animals, the metabolism of cysteine is the main source of endogenous H2S production in plants. The plants also have a characteristic sulfate reduction assimilation of H2S production. H2S synthesis occurs in chloroplasts, cytoplasm, and mitochondria [15]. First, H2S is mainly derived from cysteine degradation in the plant, catalyzed by different cysteine-degrading enzymes, including l-cysteine desulfhydrase (l-DES, EC 4.4.1.1), d-cysteine desulfhydrase (d-DES, EC 4.4.1.15), and l-3-cyanoalanine synthase (CAS, EC 4.4.1.9) [16]. Second, H2S is derived from the reductive assimilation of sulfite (SO32−) in the plants (Figure 1). These two pathways of H2S synthesis are closely linked. Sulfate or atmospheric sulfur dioxide (SO2) is the source of SO32− production in the plant in the presence of adenosine 5′-phosphosulfate (APS) reductase (APSR, EC 1.8.99.2). Atmospheric SO2 can also produce SO32− spontaneously via non-enzymatic interaction with water. Sulfite reductase (SiR, EC 1.8.7.1) reduces SO32− to H2S in the presence of chloroplast enzymes and ferredoxin [17][18]. Under alkaline conditions in the chloroplast stroma, plants spontaneously transport HS− (a dissociated form of H2S) into the cytoplasm (cytosol). With pyridoxal phosphate as a cofactor, l/d-cysteine is catalyzed by l/d-DES to produce pyruvate, NH4+, and H2S in the cytoplasm, chloroplasts, and mitochondria. Third, the nitrogenase Fe-S cluster-like (NFS/NifS-like) protein, which has similar activity to l-cysteine desulfurases, also catalyzes the conversion of cysteine to alanine and sulfur or sulfide using l-Cys as a substrate. It is also a possible source of H2S in plants [19][20]. The cyanide detoxification mechanism is also an important source of H2S in the plant (Figure 1). Ser and Acetyl-CoA are used to synthesize the intermediate reaction of O-acetyl-l-serine (OAS), catalyzed by Ser acetyltransferase (SAT, EC 2.2.1.30). O-acetyl-serine(thiol)lyase (OASTL, EC 2.5.1.47), also known as cysteine synthase, catalyzes the insertion of a particular sulfide (in this case, H2S) into the carbon skeleton via an elimination reaction, and produces cysteine [21]. CAS involves cyanide detoxification and regulates the production of H2S in mitochondria. H2S is both an intermediate reduction product of sulfate assimilation and a substrate for the synthesis of cysteine. The biosynthesis of cysteine in plastids implies a transition between a reduction in the assimilated sulfate-reducing pathway and actual metabolism [21]. The iron-sulfur cluster located in Arabidopsis mitochondria is capable of assembling the NIF system and presents cysteine desulfurase activity, which may also offer a potential source of H2S [22]. Besides the four H2S synthesis pathways described above, carbonic anhydrase (CA, EC 4.2.1.1) catalyzes the hydrolysis of COS to produce carbon dioxide (CO2) and H2S. The plants absorb COS from the air and achieve the efficient use of sulfur assimilation via CA [23][24], which may also be a source of endogenous H2S in the plant.
2. Metabolic Pathways of H2S
H2S exhibits diverse physiological and signaling roles, mainly in four distinct biochemical ways (Figure 2): (1) reacting with reactive molecule species, such as ROS, reactive nitrogen species (RNS), hypochlorite (HOCl), and reactive carbonyl species (RCS) [25][26][27]; (2) binding to the metal center of metalloproteins or the reduction of the hemoglobin center [28][29][30]; (3) post-translationally modifying proteins with specific structures (e.g., proteins containing cysteine residues (-SH)), which are mainly via S-sulfhydration [1][31][32][33]. The other PTMs are described in detail below [1]; (4) activities involving the oxidative and methylation pathways [6][34].
H2S reacts with the reactive molecule species, including ROS and RNS [1] (Figure 2). H2S can react with several biological oxidants, including superoxide radicals (O2•−), hydrogen peroxide (H2O2), hydroxyl radicals (HO·), nitrogen dioxide (NO2), peroxynitrite (ONOOH), and many others. H2S reacts readily with HOCl to form polysulfides (-S-Sn-S-) [25]. Excessive ROS and RNS levels lead to oxidative stress when plants are exposed to adversity. In turn, H2S significantly impacts the products of the plant’s defensive system. Nitric oxide (NO) is an important signaling molecule. H2S can react with NO, leading to the formation of various nitrogen (nitrous oxide (N2O), nitroxyl (HNO), S-nitrosothiols (RS-NO/SNO), and sulfur derivatives (e.g., S0, S−), which are thus involved in physiological signaling. NO converts the adversity-induced O2•− to the less toxic ONOO−. H2S further reacts with ONOO− to form thionitrate (HSNO2) [27]. This property of H2S to actively scavenge ONOO− provides strong support for the inference that it synergizes with NO to reduce ROS oxidative stress. In addition, H2S can react directly with NO to produce HNO and also react with RS-NO to form thionitrous acid (HSNO) [27]. HSNO can be metabolized to provide NO+, NO, and NO species, thus acting as a transportable NO reservoir in the organism that is involved in NO signaling.
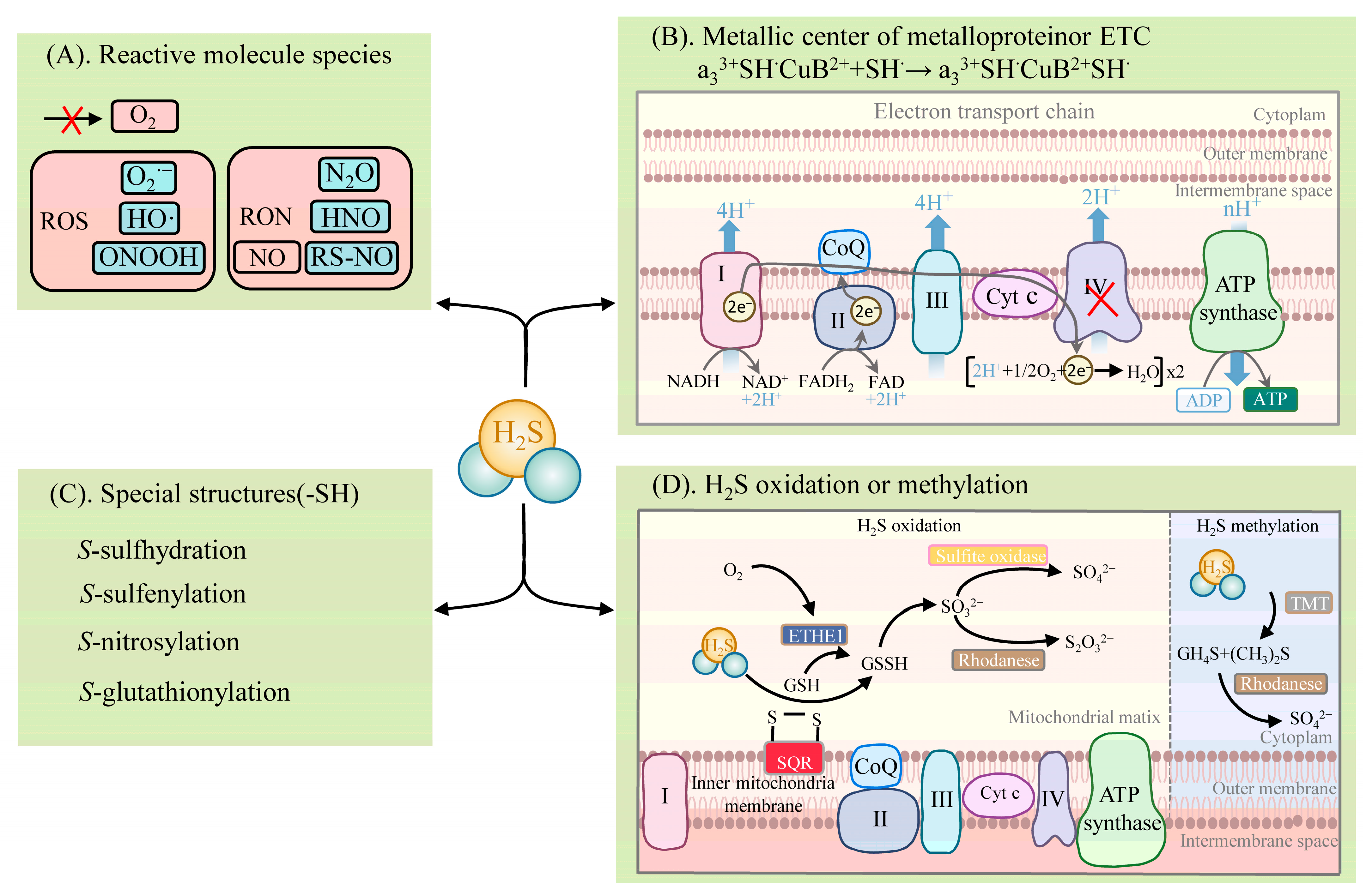
Figure 2. Metabolism of H2S: (A) reaction of H2S with reactive molecule species; (B) binding or electron transfer of H2S to the metal center of a metalloprotein; (C) reaction of H2S with proteins with specific structures (involved in post-translational modifications); (D) metabolic pathways of H2S oxidation and methylation. Note: I (mitochondrial complex I), II (mitochondrial complex II), III (mitochondrial complex III), IV (mitochondrial complex IV), Cyt c (Cytochrome c), CoQ (coenzyme Q/ubiquinone), CH4S (methanethiol), (CH3)2S (dimethyl sulfide), ETHE1 (ethylmalonic encephalopathy 1 protein), GSH (glutathione), GSSG (glutathiol), HNO (nitroxyl), H2S (hydrogen sulfide), HO· (hydroxyl radicals), N2O (nitrous oxide), NO (nitric oxide), O2 (oxygen), O2•− (superoxide radical), ONOOH (peroxynitrite), RON (reactive nitrogen species), ROS (reactive oxygen species), RS-NO (S-nitrosothiols), SO42− (sulfate), S2O32− (thiosulfuric acid), SO32− (sulfite), SQR (sulfide quinone reductase), TMT (thiol S-methyl-transferase), TST (thiosulfate sulfurtransferase). A red cross indicates an inhibitory effect on enzyme activity or the production of a substance.
H2S binds to the metal centers of metalloproteins or participates in electron transfer [1] (Figure 2). H2S involves the physiological regulation of the oxidative phosphorylation of the electron transfer chain (ETC) by means of binding to components of the ETC. It is mainly the direct binding of H2S to COX that affects ETC function, and the reduction of COX by H2S leads to the formation of HS• or S•− (which can interact with protein sulfhydryl groups (thiol)), affecting other components of the ETC. ETC complex IV, also known as COX, consists of two redox centers, Cyt a, CuA, and Cyt a3, CuB. H2S associates with the COX component, hematoxylin a3 (heme a3), and the CuB center, thus participating in the electron transfer of the ETC [28][29]. H2S favors the formation of a polar environment (tyrosine (Tyr) residues and CuB centers) around the heme a3 subunit, while H2S promotes heme a3 reduction to achieve an increase in COX enzyme activity at low concentrations. At high concentrations, H2S can bind directly to the component a3 and CuB centers of COX, resulting in the formation of the stable H2S-CuB and unstable hemoglobin H2S-Fe2+ inhibitory groups. In this case, the stability of the H2S-Fe2+ group is dependent on H2S concentration. However, the inhibition of COX by H2S can behave differently, depending on the concentration. Unlike medium concentrations, a high concentration of H2S is accompanied by the formation of stable hemoglobin a3 H2S-Fe3+ inhibitory groups, the inhibitory effect of which is irreversible [28][29][30]. In addition, the reduction of COX by H2S promotes increased ATP synthesis (which can bypass complex III to promote ETC activity) and the accumulation of reactive sulfur [6].
H2S can post-translationally modify proteins by converting the thiol group of cysteine residues to the persulfide group (-SSH) [1][31][32][33] (Figure 2). This modification is named S-sulfhydration [1]. The increased nucleophilicity of the converted persulfides, compared to the thiol group, highlights the highly reactive nature of S-sulfhydration. This also explains the potential for persulfides to act as mediators of sulfide signaling.
The metabolic pathways of H2S include oxidation and methylation [34] (Figure 2). The oxidation of H2S occurs in the mitochondria and involves several enzymes, such as sulfide quinone reductase (SQR) and the ethylmalonic encephalopathy 1 protein (ETHE1, also known as persulfide dioxygenase), thiosulfate sulfurtransferase (TST, also known as rhodanese), and mitochondrial sulfite oxidase. SQR oxidizes H2S in the inner mitochondrial membrane to produce persulfide species (e.g., glutathiol (GSSG)). At the same time, electrons released by SQR are captured by ubiquinone and transferred from H2S to coenzyme Q and to ETC at complex III. The persulfide is further oxidized by ETHE1 to produce SO32−, which is further oxidized by sulfite oxidase to SO42−, or by TST to S2O32− [34]. The metabolism of H2S by methylation occurs more as a complementary mechanism to oxidation and takes place in the cytoplasm. Then, thiol S-methyltransferase converts H2S to methanethiol (CH4S) and dimethyl sulfide (CH3)2S, which is further oxidized by rhodanese to produce thiocynate and SO42− [6].
This entry is adapted from the peer-reviewed paper 10.3390/ijms232315107
References
- Filipovic, M.R. Persulfidation (S-sulfhydration) and H2S. Handb. Exp. Pharmacol. 2015, 230, 29–59.
- Benavides, G.A.; Squadrito, G.L.; Mills, R.W.; Patel, H.D.; Isbell, T.S.; Patel, R.P.; Darley-Usmar, V.M.; Doeller, J.E.; Kraus, D.W. Hydrogen sulfide mediates the vasoactivity of garlic. Proc. Natl Acad. Sci. USA 2007, 104, 17977–17982.
- DeLeon, E.R.; Stoy, G.F.; Olson, K.R. Passive loss of hydrogen sulfide in biological experiments. Anal. Biochem. 2012, 421, 203–207.
- Powell, C.R.; Dillon, K.M.; Matson, J.B. A review of hydrogen sulfide (H2S) donors: Chemistry and potential therapeutic applications. Biochem. Pharmacol. 2018, 149, 110–123.
- Yang, J.; Minkler, P.; Grove, D.; Wang, R.; Willard, B.; Dweik, R.; Hine, C. Non-enzymatic hydrogen sulfide production from cysteine in blood is catalyzed by iron and vitamin B6. Commun. Biol. 2019, 2019, 194.
- Murphy, B.; Bhattacharya, R.; Mukherjee, P. Hydrogen sulfide signaling in mitochondria and disease. FASEB J. 2019, 33, 13098–13125.
- Kabil, O.; Banerjee, R. Enzymology of H2S biogenesis, decay and signaling. Antioxid. Redox Signal. 2014, 20, 770–782.
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Regulators of the transsulfuration pathway. Br. J. Pharmacol. 2019, 176, 583–593.
- Rose, P.; Moore, P.K.; Zhu, Y.Z. H2S biosynthesis and catabolism: New insights from molecular studies. Cell. Mol. Life Sci. 2017, 74, 1391–1412.
- Shibuya, N.; Tanaka, M.; Yoshida, M.; Ogasawara, Y.; Togawa, T.; Ishii, K.; Kimura, H. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid. Redox Signal. 2009, 11, 703–714.
- Yadav, P.K.; Yamada, K.; Chiku, T.; Koutmos, M.; Banerjee, R. Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J. Biol. Chem. 2013, 288, 20002–20013.
- Nagahara, N. Multiple role of 3-mercaptopyruvate sulfurtransferase: Antioxidative function, H2S and polysulfide production and possible SOx production. Br. J. Pharmacol. 2018, 175, 577–589.
- Kimura, H. Physiological role of hydrogen sulfide and polysulfide in the central nervous system. Neurochem. Int. 2013, 63, 492–497.
- da Costa Marques, L.A.; Teixeira, S.A.; de Jesus, F.N.; Wood, M.E.; Torregrossa, R.; Whiteman, M.; Costa, S.K.P.; Muscara, M.N. Vasorelaxant activity of AP39, a mitochondria-targeted H2S donor, on mouse mesenteric artery rings in vitro. Biomolecules 2022, 12, 280.
- Aroca, A.; Gotor, C.; Romero, L.C. Hydrogen sulfide signaling in plants: Emerging roles of protein persulfidation. Front. Plant Sci. 2018, 9, 1369.
- Youssefian, S.; Nakamura, M.; Sano, H. Tobacco plants transformed with the O-acetylserine (thiol) lyase gene of wheat are resistant to toxic levels of hydrogen sulphide gas. Plant J. Cell Mol. Biol. 1993, 4, 759–769.
- Nakayama, M.; Akashi, T.; Hase, T. Plant sulfite reductase: Molecular structure, catalytic function and interaction with ferredoxin. J. Inorg. Biochem. 2000, 82, 27–32.
- Kurmanbayeva, A.; Brychkova, G.; Bekturova, A.; Khozin, I.; Standing, D.; Yarmolinsky, D.; Sagi, M. Determination of total sulfur, sulfate, sulfite, thiosulfate, and sulfolipids in plants. Methods Mol. Biol. 2017, 1631, 253–271.
- Pilon-Smits, E.A.H.; Garifullina, G.F.; Abdel-Ghany, S.; Kato, S.-I.; Mihara, H.; Hale, K.L.; Burkhead, J.L.; Esaki, N.; Kurihara, T.; Pilon, M. Characterization of a NifS-like chloroplast protein from Arabidopsis. Implications for its role in sulfur and selenium metabolism. Plant Physiol. 2002, 130, 1309–1318.
- Hoewyk, D.V.; Pilon, M.; Pilon-Smits, E.A.H. The functions of NifS-like proteins in plant sulfur and selenium metabolism. Plant Sci. 2008, 174, 117–123.
- Wirtz, M.; Hell, R. Functional analysis of the cysteine synthase protein complex from plants: Structural, biochemical and regulatory properties. J. Plant Physiol. 2006, 163, 273–286.
- Frazzon, A.P.G.; Ramirez, M.V.; Warek, U.; Balk, J.; Frazzon, J.; Dean, D.R.; Winkel, B.S.J. Functional analysis of Arabidopsis genes involved in mitochondrial iron-sulfur cluster assembly. Plant Mol. Biol. 2007, 64, 225–240.
- Bloem, E.; Rubekin, K.; Haneklaus, S.; Banfalvi, Z.; Hesse, H.; Schnug, E. H2S and COS gas exchange of transgenic potato lines with modified expression levels of enzymes involved in sulphur metabolism. J. Agron. Crop Sci. 2011, 197, 311–321.
- Yamasaki, H.; Cohen, M.F. Biological consilience of hydrogen sulfide and nitric oxide in plants: Gases of primordial earth linking plant, microbial and animal physiologies. Nitric Oxide 2016, 55–56, 91–100.
- Nagy, P.; Winterbourn, C.C. Rapid reaction of hydrogen sulfide with the neutrophil oxidant hypochlorous acid to generate polysulfides. Chem. Res. Toxicol. 2010, 23, 1541–1543.
- Sun, H.J.; Wu, Z.Y.; Cao, L.; Zhu, M.Y.; Nie, X.W.; Huang, D.J.; Sun, M.T.; Bian, J.S. Role of nitroxyl (HNO) in cardiovascular system: From biochemistry to pharmacology. Pharmacol. Res. 2020, 159, 10.
- Filipovic, M.R.; Miljkovic, J.L.; Nauser, T.; Royzen, M.; Klos, K.; Shubina, T.; Koppenol, W.H.; Lippard, S.J.; Ivanovic-Burmazovic, I. Chemical characterization of the smallest S-nitrosothiol, HSNO; cellular cross-talk of H2S and S-nitrosothiols. J. Am. Chem. Soc. 2012, 134, 12016–12027.
- Nicholls, P.; Marshall, D.C.; Cooper, C.E.; Wilson, M.T. Sulfide inhibition of and metabolism by cytochrome c oxidase. Biochem. Soc. Trans. 2013, 41, 1312–1316.
- Hill, B.C.; Woon, T.C.; Nicholls, P.; Peterson, J.; Greenwood, C.; Thomson, A.J. Interactions of sulphide and other ligands with cytochrome c oxidase. An electron-paramagnetic-resonance study. Biochem. J. 1984, 224, 591–600.
- Modis, K.; Coletta, C.; Erdelyi, K.; Papapetropoulos, A.; Szabo, C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013, 27, 601–611.
- Shen, J.; Zhang, J.; Zhou, M.; Zhou, H.; Cui, B.; Gotor, C.; Romero, L.C.; Fu, L.; Yang, J.; Foyer, C.H.; et al. Persulfidation-based modification of cysteine desulfhydrase and the NADPH oxidase RBOHD controls guard cell abscisic acid signaling. Plant Cell 2020, 32, 1000–1017.
- Zhou, M.; Zhang, J.; Shen, J.; Zhou, H.; Zhao, D.; Gotor, C.; Romero, L.C.; Fu, L.; Li, Z.; Yang, J.; et al. Hydrogen sulfide-linked persulfidation of ABI4 controls ABA responses through the transactivation of MAPKKK18 in Arabidopsis. Mol. Plant. 2021, 14, 921–936.
- Corpas, F.J.; Gonzalez-Gordo, S.; Palma, J.M. Nitric oxide and hydrogen sulfide modulate the NADPH-generating enzymatic system in higher plants. J. Exp. Bot. 2021, 72, 830–847.
- Gerush, I.V.; Ferenchuk, Y.O. Hydrogen sulfide and mitochondria. Biopolym. Cell 2019, 35, 3–15.
This entry is offline, you can click here to edit this entry!