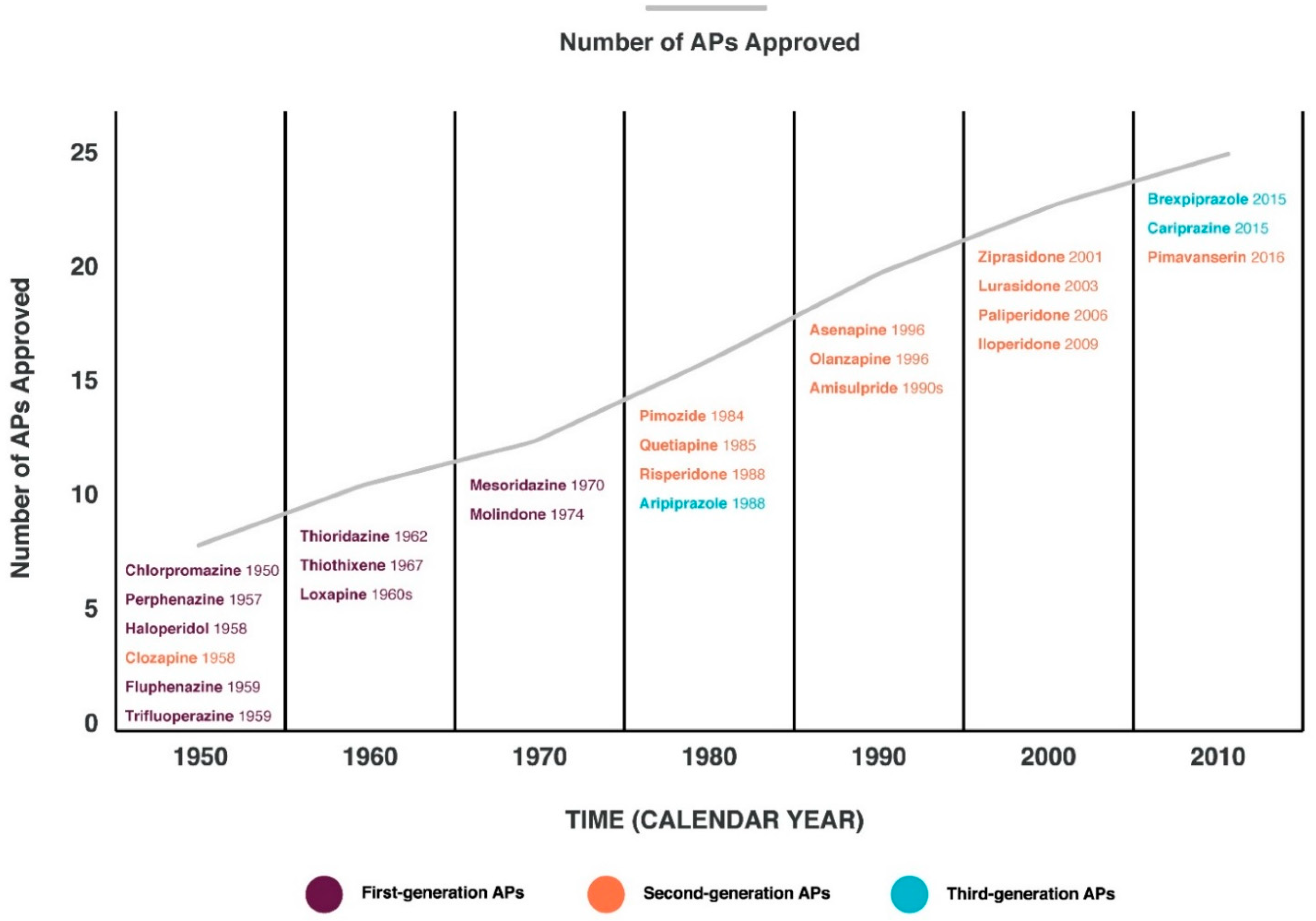
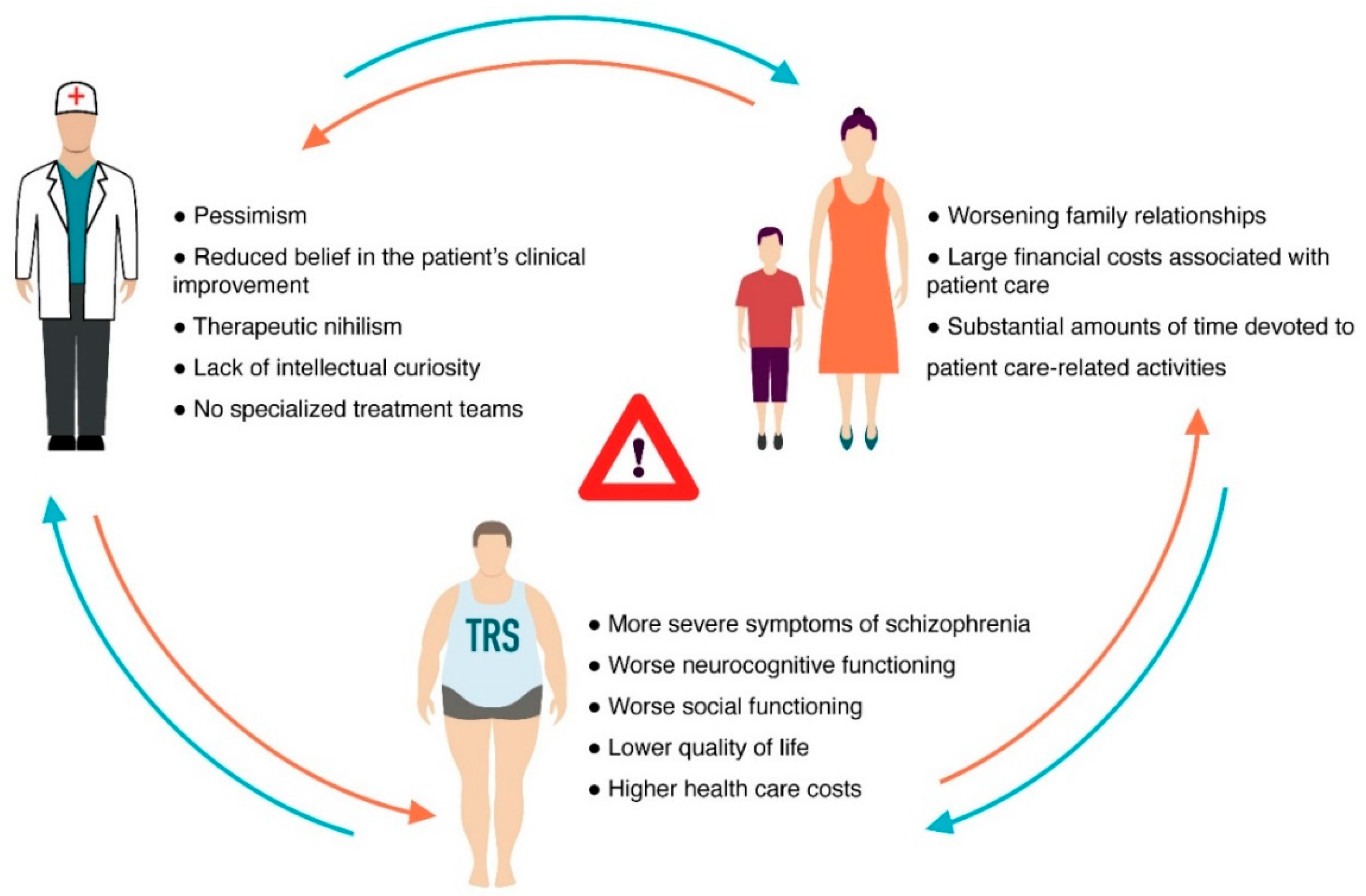
Schizophrenia (Sch) is a severe and widespread mental disorder. Antipsychotics (APs) of the first and new generations as the first-line treatment of Sch are not effective in about a third of cases and are also unable to treat negative symptoms and cognitive deficits of schizophrenics. This explains the search for new therapeutic strategies for a disease-modifying therapy for treatment-resistant Sch (TRS). Biological compounds are of great interest to researchers and clinicians, among which D-Serine (D-Ser) and D-Aspartate (D-Asp) are among the promising ones. The Sch glutamate theory suggests that neurotransmission dysfunction caused by glutamate N-methyl-D-aspartate receptors (NMDARs) may represent a primary deficiency in this mental disorder and play an important role in the development of TRS. D-Ser and D-Asp are direct NMDAR agonists and may be involved in modulating the functional activity of dopaminergic neurons.
- D-serine
- D-aspartate
- disease-modifying therapy
- treatment-resistant schizophrenia
1. Introduction
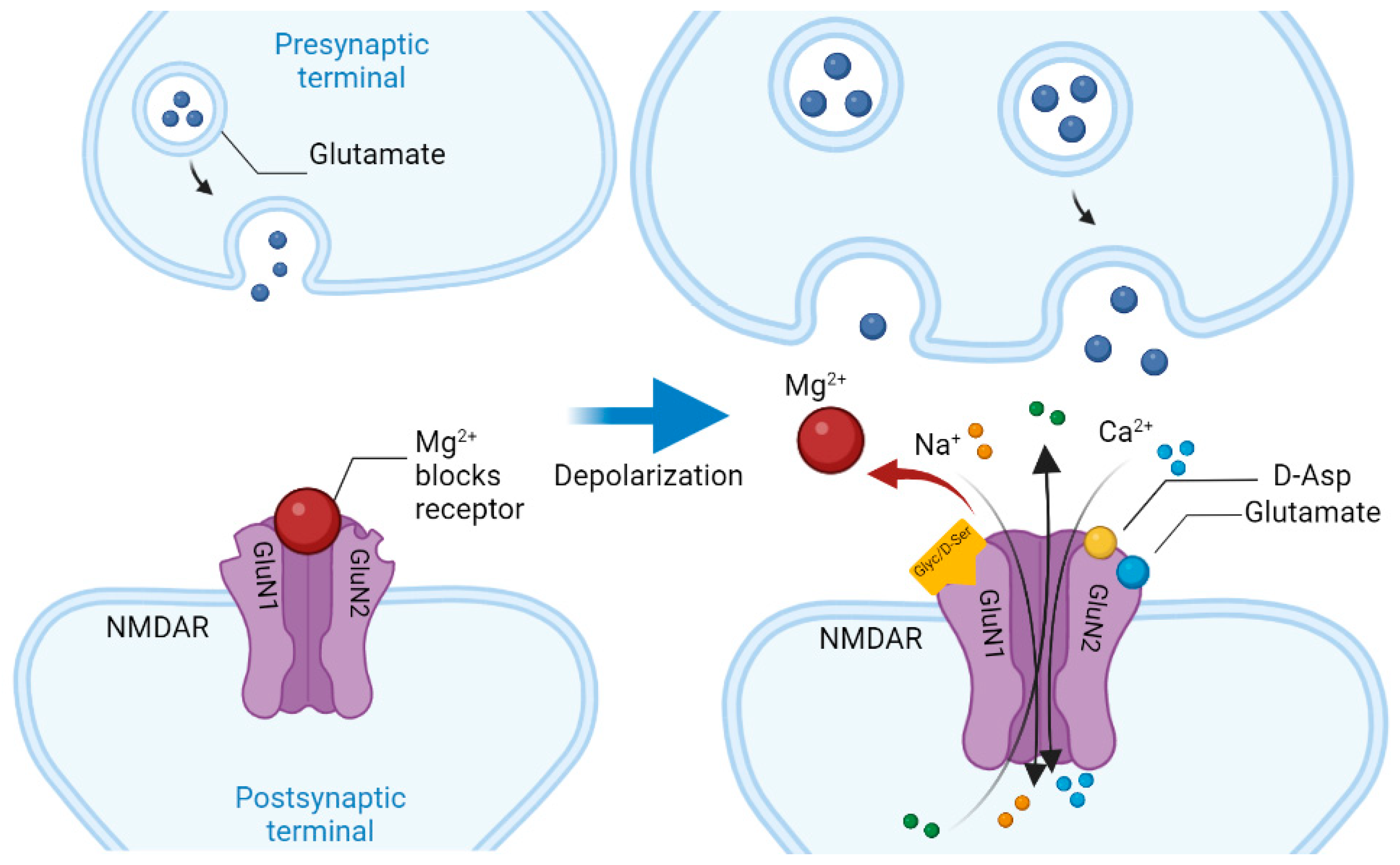
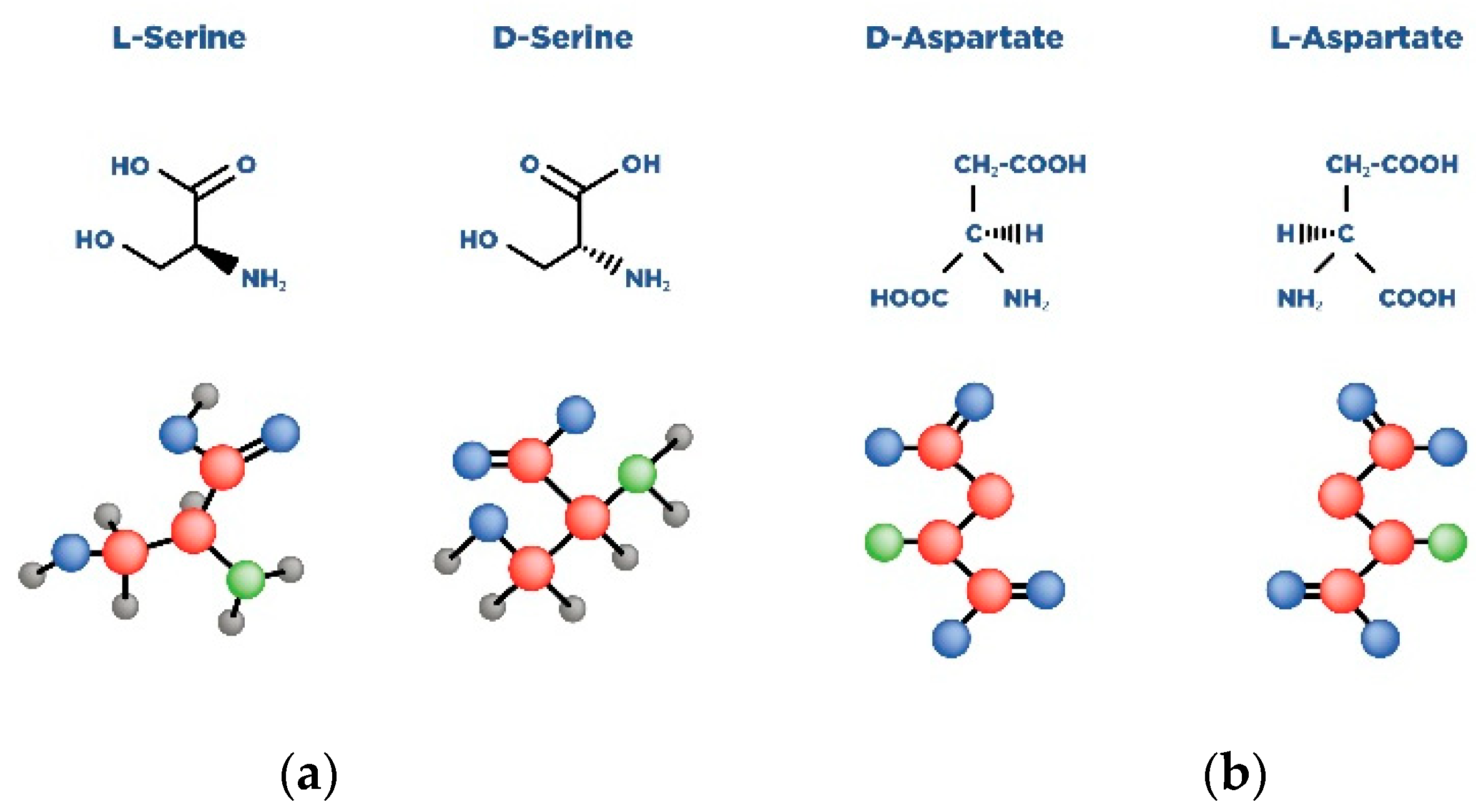
| Amino Acid | Receptor | Function |
|---|---|---|
| D-aspartate | Synaptic NMDAR | Binds to the L-Glut region of ionotropic NMDARs. |
| D-serine | Synaptic NMDAR | A full agonist of the NMDAR Glyc modulatory site. Physiological ligand of the co-agonist site. Reduces NMDAR-mediated transmission in vitro via non-vesicular release via ASC-1 transporters. |
2. D-Aspartate
2.1. The Biological Role of D-Aspartate
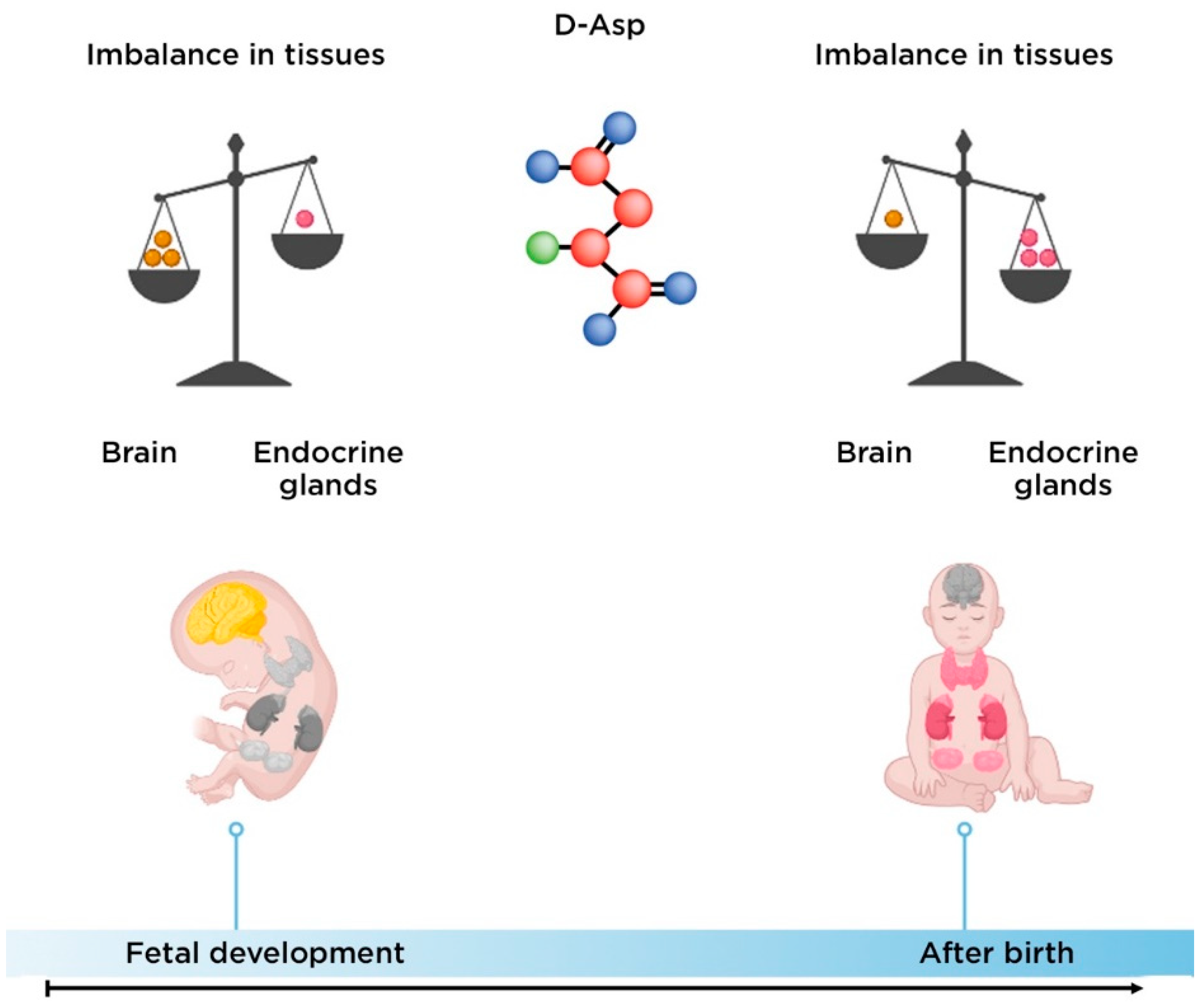
2.2. The Role of D-Aspartate in the Pathogenesis of Schizophrenia
3. D-Serine
3.1. Biological Role of D-Serine
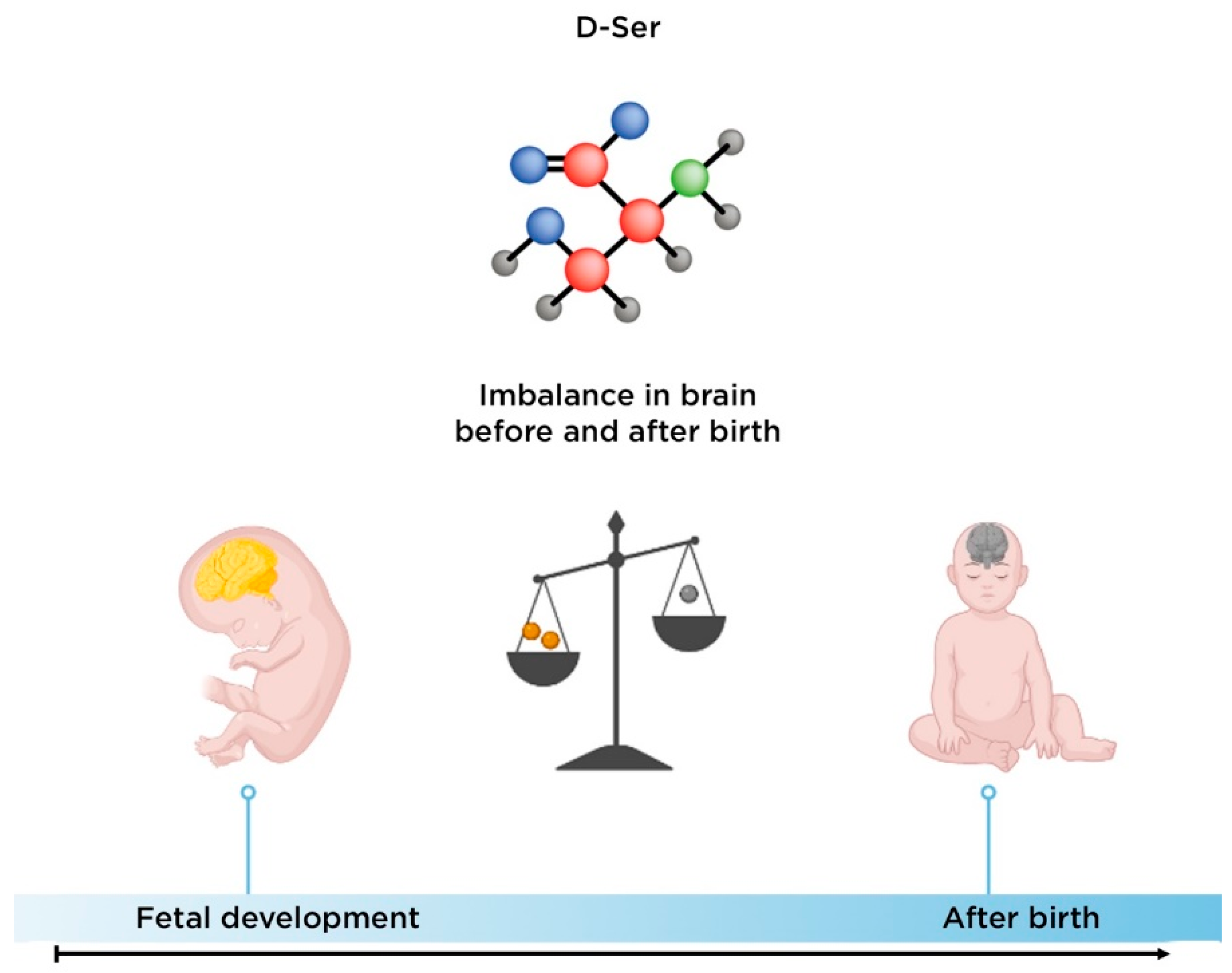
3.2. The Role of D-Serine in the Development of Schizophrenia
This entry is adapted from the peer-reviewed paper 10.3390/nu14235142
References
- Charlson, F.J.; Ferrari, A.J.; Santomauro, D.F.; Diminic, S.; Stockings, E.; Scott, J.G.; McGrath, J.J.; Whiteford, H.A. Global epidemiology and burden of schizophrenia: Findings from the global burden of disease study 2016. Schizophr. Bull. 2018, 44, 1195–1203.
- Vita, A.; Minelli, A.; Barlati, S.; Deste, G.; Giacopuzzi, E.; Valsecchi, P.; Turrina, C.; Gennarelli, M. Treatment-resistant schizophrenia: Genetic and neuroimaging correlates. Front. Pharmacol. 2019, 10, 402.
- Howes, O.D.; Shatalina, E. Integrating the neurodevelopmental and dopamine hypotheses of schizophrenia and the role of cortical excitation-inhibition balance. Biol. Psychiatry 2022, 92, 501–513.
- Plitman, E.; Iwata, Y.; Caravaggio, F.; Nakajima, S.; Chung, J.K.; Gerretsen, P.; Kim, J.; Takeuchi, H.; Chakravarty, M.M.; Remington, G.; et al. Kynurenic acid in schizophrenia: A systematic review and meta-analysis. Schizophr. Bull. 2017, 43, 764–777.
- Uno, Y.; Coyle, J.T. Glutamate hypothesis in schizophrenia. Psychiatry Clin. Neurosci. 2019, 73, 204–215.
- Adell, A. Brain NMDA receptors in schizophrenia and depression. Biomolecules 2020, 10, 947.
- Seeman, P.; Kapur, S. Schizophrenia: More dopamine, more D2 receptors. Proc. Natl. Acad. Sci. USA 2000, 97, 7673–7675.
- Nasyrova, R.F.; Neznanov, N.G. Clinical Psychopharmacogenetics, 1st ed.; DEAN Publishing House: Saint Petersburg, Russia, 2019; pp. 93–174. (In Russian)
- Shnayder, N.A.; Khasanova, A.K.; Strelnik, A.I.; Al-Zamil, M.; Otmakhov, A.P.; Neznanov, N.G.; Shipulin, G.A.; Petrova, M.M.; Garganeeva, N.P.; Nasyrova, R.F. Cytokine imbalance as a biomarker of treatment-resistant schizophrenia. Int. J. Mol. Sci. 2022, 23, 11324.
- de Bartolomeis, A.; Errico, F.; Aceto, G.; Tomasetti, C.; Usiello, A.; Iasevoli, F. D-aspartate dysregulation in Ddo (−/−) mice modulates phencyclidine-induced gene expression changes of postsynaptic density molecules in cortex and striatum. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2015, 62, 35–43.
- : FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm (accessed on 20 September 2022).
- Wagner, E.; Kane, J.M.; Correll, C.U.; Howes, O.; Siskind, D.; Honer, W.G.; Lee, J.; Falkai, P.; Schneider-Axmann, T.; Hasan, A.; et al. Clozapine combination and augmentation strategies in patients with schizophrenia -recommendations from an international expert survey among the treatment response and resistance in psychosis (TRRIP) working group. Schizophr. Bull. 2020, 46, 1459–1470.
- Polese, D.; Fornaro, M.; Palermo, M.; De Luca, V.; de Bartolomeis, A. Treatment-resistant to antipsychotics: A resistance to everything? psychotherapy in treatment-resistant schizophrenia and nonaffective psychosis: A 25-year systematic review and exploratory meta-analysis. Front. Psychiatry 2019, 10, 210.
- Kane, J.; Honigfeld, G.; Singer, J.; Meltzer, H. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch. Gen. Psychiatry 1988, 45, 789–796.
- Ajnakina, O.; Horsdal, H.T.; Lally, J.; MacCabe, J.H.; Murray, R.M.; Gasse, C.; Wimberley, T. Validation of an algorithm-based definition of treatment resistance in patients with schizophrenia. Schizophr. Res. 2018, 197, 294–297.
- Elkis, H.; Buckley, P.F. Treatment-Resistant Schizophrenia. Psychiatr. Clin. N. Am. 2016, 39, 239–265.
- Remington, G.; Addington, D.; Honer, W.; Ismail, Z.; Raedler, T.; Teehan, M. Guidelines for the pharmacotherapy of schizophrenia in adults. Can. J. Psychiatry 2017, 62, 604–616.
- Silverstein, S.M.; Bellack, A.S. A scientific agenda for the concept of recovery as it applies to schizophrenia. Clin. Psychol. Rev. 2008, 28, 1108–1124.
- Kennedy, J.L.; Altar, C.A.; Taylor, D.L.; Degtiar, I.; Hornberger, J.C. The social and economic burden of treatment-resistant schizophrenia: A systematic literature review. Int. Clin. Psychopharmacol. 2014, 29, 63–76.
- Lally, J.; Ajnakina, O.; Di Forti, M.; Trotta, A.; Demjaha, A.; Kolliakou, A.; Mondelli, V.; Reis Marques, T.; Pariante, C.; Dazzan, P.; et al. Two distinct patterns of treatment resistance: Clinical predictors of treatment resistance in first-episode schizophrenia spectrum psychoses. Psychol. Med. 2016, 46, 3231–3240.
- Sheitman, B.B.; Lieberman, J.A. The natural history and pathophysiology of treatment resistant schizophrenia. J. Psychiatr. Res. 1998, 32, 143–150.
- Stępnicki, P.; Kondej, M.; Kaczor, A.A. Current concepts and treatments of schizophrenia. Molecules 2018, 23, 2087.
- Bendikov, I.; Nadri, C.; Amar, S.; Panizzutti, R.; De Miranda, J.; Wolosker, H.; Agam, G. A CSF and postmortem brain study of D-serine metabolic parameters in schizophrenia. Schizophr. Res. 2007, 90, 41–51.
- Teo, C.; Zai, C.; Borlido, C.; Tomasetti, C.; Strauss, J.; Shinkai, T.; Le Foll, B.; Wong, A.; Kennedy, J.L.; De Luca, V. Analysis of treatment-resistant schizophrenia and 384 markers from candidate genes. Pharm. Genom. 2012, 22, 807–811.
- Shen, L.; Lv, X.; Huang, H.; Li, M.; Huai, C.; Wu, X.; Wu, H.; Ma, J.; Chen, L.; Wang, T.; et al. Genome-wide analysis of DNA methylation in 106 schizophrenia family trios in Han Chinese. eBioMedicine 2021, 72, 103609.
- Wagh, V.V.; Vyas, P.; Agrawal, S.; Pachpor, T.A.; Paralikar, V.; Khare, S.P. Peripheral blood-based gene expression studies in schizophrenia: A systematic review. Front. Genet. 2021, 12, 736483.
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427.
- Glasgow, N.G.; Siegler Retchless, B.; Johnson, J.W. Molecular bases of NMDA receptor subtype-dependent properties. J. Physiol. 2015, 593, 83–95.
- Vieira, M.; Yong, X.; Roche, K.W.; Anggono, V. Regulation of NMDA glutamate receptor functions by the GluN2 subunits. J. Neurochem. 2020, 154, 121–143.
- Radulovic, J.; Ren, L.Y.; Gao, C. N-Methyl D-aspartate receptor subunit signaling in fear extinction. Psychopharmacology 2019, 236, 239–250.
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414.
- Krashia, P.; Ledonne, A.; Nobili, A.; Cordella, A.; Errico, F.; Usiello, A.; D′Amelio, M.; Mercuri, N.B.; Guatteo, E.; Carunchio, I. Persistent elevation of D-Aspartate enhances NMDA receptor-mediated responses in mouse substantia nigra pars compacta dopamine neurons. Neuropharmacology 2016, 103, 69–78.
- Cristino, L.; Luongo, L.; Squillace, M.; Paolone, G.; Mango, D.; Piccinin, S.; Zianni, E.; Imperatore, R.; Iannotta, M.; Longo, F.; et al. d-Aspartate oxidase influences glutamatergic system homeostasis in mammalian brain. Neurobiol. Aging 2015, 36, 1890–1902.
- Errico, F.; Mothet, J.P.; Usiello, A. D-aspartate: An endogenous NMDA receptor agonist enriched in the developing brain with potential involvement in schizophrenia. J. Pharm. Biomed. Anal. 2015, 116, 7–17.
- Errico, F.; Nisticò, R.; Di Giorgio, A.; Squillace, M.; Vitucci, D.; Galbusera, A.; Piccinin, S.; Mango, D.; Fazio, L.; Middei, S.; et al. Free D-aspartate regulates neuronal dendritic morphology, synaptic plasticity, gray matter volume and brain activity in mammals. Transl. Psychiatry 2014, 4, e417.
- Abdulbagi, M.; Wang, L.; Siddig, O.; Di, B.; Li, B. D-amino acids and d-amino acid-containing peptides: Potential disease biomarkers and therapeutic targets? Biomolecules 2021, 11, 1716.
- Biomolecules, Science: Amino Acids: Building Blocks of Proteins. Available online: https://conductscience.com/amino-acids-building-blocks-of-proteins/ (accessed on 20 September 2022).
- de Bartolomeis, A.; Vellucci, L.; Austin, M.C.; De Simone, G.; Barone, A. Rational and translational implications of d-amino acids for treatment-resistant schizophrenia: From neurobiology to the clinics. Biomolecules 2022, 12, 909.
- Billard, J.M. D-Amino acids in brain neurotransmission and synaptic plasticity. Amino Acids 2012, 43, 1851–1860.
- Balu, D.T. The NMDA receptor and schizophrenia: From pathophysiology to treatment. Adv. Pharmacol. 2016, 76, 351–382.
- Jiménez-Sánchez, L.; Campa, L.; Auberson, Y.P.; Adell, A. The role of GluN2A and GluN2B subunits on the effects of NMDA receptor antagonists in modeling schizophrenia and treating refractory depression. Neuropsychopharmacology 2014, 39, 2673–2680.
- Metabocard for D-Aspartic Acid (HMDB0006483). Human Metabolome Database. Available online: https://hmdb.ca/metabolites/HMDB0006483 (accessed on 20 September 2022).
- Kiriyama, Y.; Nochi, H. D-amino acids in the nervous and endocrine systems. Scientifica 2016, 2016, 6494621.
- Liang, R.; Robb, F.T.; Onstott, T.C. Aspartic acid racemization and repair in the survival and recovery of hyperthermophiles after prolonged starvation at high temperature. FEMS Microbiol. Ecol. 2021, 97, fiab112.
- Bastings, J.; van Eijk, H.M.; Olde Damink, S.W.; Rensen, S.S. D-amino acids in health and disease: A focus on cancer. Nutrients 2019, 11, 2205.
- Usiello, A.; Di Fiore, M.M.; De Rosa, A.; Falvo, S.; Errico, F.; Santillo, A.; Nuzzo, T.; Chieffi Baccari, G. New evidence on the role of d-aspartate metabolism in regulating brain and endocrine system physiology: From preclinical observations to clinical applications. Int. J. Mol. Sci. 2020, 21, 8718.
- Errico, F.; Napolitano, F.; Nisticò, R.; Usiello, A. New insights on the role of free d-aspartate in the mammalian brain. Amino Acids 2012, 43, 1861–1871.
- Di Fiore, M.M.; Santillo, A.; Chieffi Baccari, G. Current knowledge of d-aspartate in glandular tissues. Amino Acids 2014, 46, 1805–1818.
- Li, Y.; Han, H.; Yin, J.; Li, T.; Yin, Y. Role of D-aspartate on biosynthesis, racemization, and potential functions: A mini-review. Anim. Nutr. 2018, 4, 311–315.
- D′Aniello, A.; Guiditta, A. Identification of d-aspartic acid in the brain of octopus vulgaris lam. J. Neurochem. 1977, 29, 1053–1057.
- D′Aniello, G.; Ronsini, S.; Guida, F.; Spinelli, P.; D′Aniello, A. Occurrence of D-aspartic acid in human seminal plasma and spermatozoa: Possible role in reproduction. Fertil. Steril. 2005, 84, 1444–1449.
- D′Aniello, G.; Grieco, N.; Di Filippo, M.A.; Cappiello, F.; Topo, E.; D′Aniello, E.; Ronsini, S. Reproductive implication of D-aspartic acid in human pre-ovulatory follicular fluid. Hum. Reprod. 2007, 22, 3178–3183.
- Ota, N.; Shi, T.; Sweedler, J.V. D-Aspartate acts as a signaling molecule in nervous and neuroendocrine systems. Amino Acids 2012, 43, 1873–1886.
- Wolosker, H.; D′Aniello, A.; Snyder, S.H. D-aspartate disposition in neuronal and endocrine tissues: Ontogeny, biosynthesis and release. Neuroscience 2000, 100, 183–189.
- Hons, J.; Zirko, R.; Ulrychova, M.; Cermakova, E.; Doubek, P.; Libiger, J. Glycine serum level in schizophrenia: Relation to negative symptoms. Psychiatry Res. 2010, 176, 103–108.
- Singh, S.P.; Singh, V. Meta-analysis of the efficacy of adjunctive NMDA receptor modulators in chronic schizophrenia. CNS Drugs 2011, 25, 859–885.
- Sumiyoshi, T.; Anil, A.E.; Jin, D.; Jayathilake, K.; Lee, M.; Meltzer, H.Y. Plasma glycine and serine levels in schizophrenia compared to normal controls and major depression: Relation to negative symptoms. Int. J. Neuropsychopharmacol. 2004, 7, 1–8.
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Clozapine, but not haloperidol, enhances glial D-serine and L-glutamate release in rat frontal cortex and primary cultured astrocytes. Br. J. Pharmacol. 2012, 165, 1543–1555.
- Tsai, G.E.; Yang, P.; Chung, L.C.; Tsai, I.C.; Tsai, C.W.; Coyle, J.T. D-serine added to clozapine for the treatment of schizophrenia. Am. J. Psychiatry 1999, 156, 1822–1825.
- Tsai, G.E.; Lin, P.Y. Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia, a critical review and meta-analysis. Curr. Pharm. Des. 2010, 16, 522–537.
- Verrall, L.; Walker, M.; Rawlings, N.; Benzel, I.; Kew, J.N.; Harrison, P.J.; Burnet, P.W. D-amino acid oxidase and serine racemase in human brain: Normal distribution and altered expression in schizophrenia. Eur. J. Neurosci. 2007, 26, 1657–1669.
- Gong, X.Q.; Frandsen, A.; Lu, W.Y.; Wan, Y.; Zabek, R.L.; Pickering, D.S.; Bai, D. D-aspartate and NMDA, but not L-aspartate, block AMPA receptors in rat hippocampal neurons. Br. J. Pharm. 2005, 145, 449–459.
- Errico, F.; Nuzzo, T.; Carella, M.; Bertolino, A.; Usiello, A. The emerging role of altered d-aspartate metabolism in schizophrenia: New insights from preclinical models and human studies. Front. Psychiatry 2018, 9, 559.
- Weiser, M.; Heresco-Levy, U.; Davidson, M.; Javitt, D.C.; Werbeloff, N.; Gershon, A.A.; Abramovich, Y.; Amital, D.; Doron, A.; Konas, S.; et al. A multicenter, add-on randomized controlled trial of low-dose d-serine for negative and cognitive symptoms of schizophrenia. J. Clin. Psychiatry 2012, 73, e728–e734.
- Kantrowitz, J.T.; Malhotra, A.K.; Cornblatt, B.; Silipo, G.; Balla, A.; Suckow, R.F.; D′Souza, C.; Saksa, J.; Woods, S.W.; Javitt, D.C. High dose D-serine in the treatment of schizophrenia. Schizophr. Res. 2010, 121, 125–130.
- Wolosker, H.; Sheth, K.N.; Takahashi, M.; Mothet, J.P.; Brady, R.O.; Ferris, C.D., Jr.; Snyder, S.H. Purification of serine racemase: Biosynthesis of the neuromodulator D-serine. Proc. Natl. Acad. Sci. USA 1999, 96, 721–725.
- Yamada, K.; Ohnishi, T.; Hashimoto, K.; Ohba, H.; Iwayama-Shigeno, Y.; Toyoshima, M.; Okuno, A.; Takao, H.; Toyota, T.; Minabe, Y.; et al. Identification of multiple serine racemase (SRR) mRNA isoforms and genetic analyses of SRR and DAO in schizophrenia and D-serine levels. Biol. Psychiatry 2005, 57, 1493–1503.
- Yamamori, H.; Hashimoto, R.; Fujita, Y.; Numata, S.; Yasuda, Y.; Fujimoto, M.; Ohi, K.; Umeda-Yano, S.; Ito, A.; Ohmori, T.; et al. Changes in plasma D-serine, L-serine, and glycine levels in treatment-resistant schizophrenia before and after clozapine treatment. Neurosci. Lett. 2014, 582, 93–98.
- Assisi, L.; Botte, V.; D′Aniello, A.; Di Fiore, M.M. Enhancement of aromatase activity by D-aspartic acid in the ovary of the lizard Podarcis s. sicula. Reproduction 2001, 121, 803–808.
- Bi, C.; Zheng, X.; Azaria, S.; Beeram, S.; Li, Z.; Hage, D.S. Chromatographic studies of protein-based chiral separations. Separations 2016, 3, 27.
- Kantrowitz, J.T.; Epstein, M.L.; Lee, M.; Lehrfeld, N.; Nolan, K.A.; Shope, C.; Petkova, E.; Silipo, G.; Javitt, D.C. Improvement in mismatch negativity generation during d-serine treatment in schizophrenia: Correlation with symptoms. Schizophr. Res. 2018, 191, 70–79.
- Dunlop, D.S.; Neidle, A.; McHale, D.; Dunlop, D.M.; Lajtha, A. The presence of free D-aspartic acid in rodents and man. Biochem. Biophys. Res. Commun. 1986, 141, 27–32.
- Beneyto, M.; Kristiansen, L.V.; Oni-Orisan, A.; McCullumsmith, R.E.; Meador-Woodruff, J.H. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology 2007, 32, 1888–1902.
- Choi, K.H.; Wykes, T.; Kurtz, M.M. Adjunctive pharmacotherapy for cognitive deficits in schizophrenia: Meta-analytical investigation of efficacy. Br. J. Psychiatry 2013, 203, 172–178.
- Olney, J.W.; Farber, N.B. Glutamate receptor dysfunction and schizophrenia. Arch. Gen. Psychiatry 1995, 52, 998–1007.
- Wu, Q.; Huang, J.; Wu, R. Drugs Based on NMDAR Hypofunction Hypothesis in Schizophrenia. Front. Neurosci. 2021, 15, 641047.
- D’Aniello, A. D-Aspartic acid: An endogenous amino acid with an important neuroendocrine role. Brain Res. Rev. 2007, 53, 215–234.
- Errico, F.; Rossi, S.; Napolitano, F.; Catuogno, V.; Topo, E.; Fisone, G.; D′Aniello, A.; Centonze, D.; Usiello, A. D-aspartate prevents corticostriatal long-term depression and attenuates schizophrenia-like symptoms induced by amphetamine and MK-801. J. Neurosci. 2008, 28, 10404–10414.
- Errico, F.; D′Argenio, V.; Sforazzini, F.; Iasevoli, F.; Squillace, M.; Guerri, G.; Napolitano, F.; Angrisano, T.; Di Maio, A.; Keller, S.; et al. A role for D-aspartate oxidase in schizophrenia and in schizophrenia-related symptoms induced by phencyclidine in mice. Transl. Psychiatry 2015, 5, e512.
- Zhou, Y.; Shu, N.; Liu, Y.; Song, M.; Hao, Y.; Liu, H.; Yu, C.; Liu, Z.; Jiang, T. Altered resting-state functional connectivity and anatomical connectivity of hippocampus in schizophrenia. Schizophr. Res. 2008, 100, 120–132.
- Kitamura, A.; Hojo, Y.; Ikeda, M.; Karakawa, S.; Kuwahara, T.; Kim, J.; Soma, M.; Kawato, S.; Tsurugizawa, T. Ingested d-aspartate facilitates the functional connectivity and modifies dendritic spine morphology in rat hippocampus. Cereb. Cortex 2019, 29, 2499–2508.
- Metabocard for D-Serine (HMDB0003406). Human Metabolome Database. Available online: https://hmdb.ca/metabolites/HMDB0003406 (accessed on 20 September 2022).
- Ito, T.; Hamauchi, N.; Hagi, T.; Morohashi, N.; Hemmi, H.; Sato, Y.G.; Saito, T.; Yoshimura, T. D-serine metabolism and its importance in development of dictyostelium discoideum. Front. Microbiol. 2018, 9, 784.
- Hashimoto, A.; Kumashiro, S.; Nishikawa, T.; Oka, T.; Takahashi, K.; Mito, T.; Takashima, S.; Doi, N.; Mizutani, Y.; Yamazaki, T. Embryonic development and postnatal changes in free D-aspartate and D-serine in the human prefrontal cortex. J. Neurochem. 1993, 61, 348–351.
- Kantrowitz, J.T.; Woods, S.W.; Petkova, E.; Cornblatt, B.; Corcoran, C.M.; Chen, H.; Silipo, G.; Javitt, D.C. D-serine for the treatment of negative symptoms in individuals at clinical high risk of schizophrenia: A pilot, double-blind, placebo-controlled, randomised parallel group mechanistic proof-of-concept trial. Lancet Psychiatry 2015, 2, 403–412.
- Yang, S.; Qiao, H.; Wen, L.; Zhou, W.; Zhang, Y. D-serine enhances impaired long-term potentiation in CA1 subfield of hippocampal slices from aged senescence-accelerated mouse prone/8. Neurosci. Lett. 2005, 379, 7–12.
- Yang, Y.; Ge, W.; Chen, Y.; Zhang, Z.; Shen, W.; Wu, C.; Poo, M.; Duan, S. Contribution of astrocytes to hippocampal long-term potentiation through release of D-serine. Proc. Natl. Acad. Sci. USA 2003, 100, 15194–15199.
- Shimazaki, T.; Kaku, A.; Chaki, S. D-Serine and a glycine transporter-1 inhibitor enhance social memory in rats. Psychopharmacology 2010, 209, 263–270.
- Durrant, A.R.; Heresco-Levy, U. D-Serine in neuropsychiatric disorders: New advances. Adv. Psychiatry 2014, 2014, 1–16.
- Krystal, J.H.; Karper, L.P.; Seibyl, J.P.; Freeman, G.K.; Delaney, R.; Bremner, J.D.; Heninger, G.R.; Bowers, M.B.; Charney, D.S., Jr. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 1994, 51, 199–214.
- Fujita, Y.; Ishima, T.; Hashimoto, K. Supplementation with D-serine prevents the onset of cognitive deficits in adult offspring after maternal immune activation. Sci. Rep. 2016, 6, 37261.
- Andersen, J.; Pouzet, B. Spatial Memory Deficits Induced by Perinatal Treatment of Rats with PCP and Reversal Effect of D-Serine. Neuropsychopharmacology 2004, 29, 1080–1090.
- MacKay, M.B.; Kravtsenyuk, M.; Thomas, R.; Mitchell, N.D.; Dursun, S.M.; Baker, G.B. D-serine: Potential therapeutic agent and/or biomarker in schizophrenia and depression? Front. Psychiatry 2019, 10, 25.
- Taniguchi, K.; Sawamura, H.; Ikeda, Y.; Tsuji, A.; Kitagishi, Y.; Matsuda, S. D-amino acids as a biomarker in schizophrenia. Diseases 2022, 10, 9.
- Hons, J.; Zirko, R.; Vasatova, M.; Doubek, P.; Klimova, B.; Masopust, J.; Valis, M.; Kuca, K. Impairment of executive functions associated with lower d-serine serum levels in patients with schizophrenia. Front. Psychiatry 2021, 12, 514579.
- Hashimoto, K.; Fukushima, T.; Shimizu, E.; Komatsu, N.; Watanabe, H.; Shinoda, N.; Nakazato, M.; Kumakiri, C.; Okada, S.; Hasegawa, H.; et al. Decreased serum levels of D-serine in patients with schizophrenia: Evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch. Gen. Psychiatry 2003, 60, 572–576.
- Balu, D.T.; Li, Y.; Puhl, M.D.; Benneyworth, M.A.; Basu, A.C.; Takagi, S.; Bolshakov, V.Y.; Coyle, J.T. Multiple risk pathways for schizophrenia converge in serine racemase knockout mice, a mouse model of NMDA receptor hypofunction. Proc. Natl. Acad. Sci. USA 2013, 110, E2400–E2409.
- Habl, G.; Zink, M.; Petroianu, G.; Bauer, M.; Schneider-Axmann, T.; von Wilmsdorff, M.; Falkai, P.; Henn, F.A.; Schmitt, A. Increased D-amino acid oxidase expression in the bilateral hippocampal CA4 of schizophrenic patients: A post-mortem study. J. Neural Transm. 2009, 116, 1657–1665.
- Burnet, P.W.; Eastwood, S.L.; Bristow, G.C.; Godlewska, B.R.; Sikka, P.; Walker, M.; Harrison, P.J. D-amino acid oxidase activity and expression are increased in schizophrenia. Mol. Psychiatry 2008, 13, 658–660.
- Phillips, J.R.; Hewedi, D.H.; Eissa, A.M.; Moustafa, A.A. The cerebellum and psychiatric disorders. Front. Public Health 2015, 3, 66.
- Labrie, V.; Fukumura, R.; Rastogi, A.; Fick, L.J.; Wang, W.; Boutros, P.C.; Kennedy, J.L.; Semeralul, M.O.; Lee, F.H.; Baker, G.B.; et al. Serine racemase is associated with schizophrenia susceptibility in humans and in a mouse model. Hum. Mol. Genet. 2009, 18, 3227–3243.