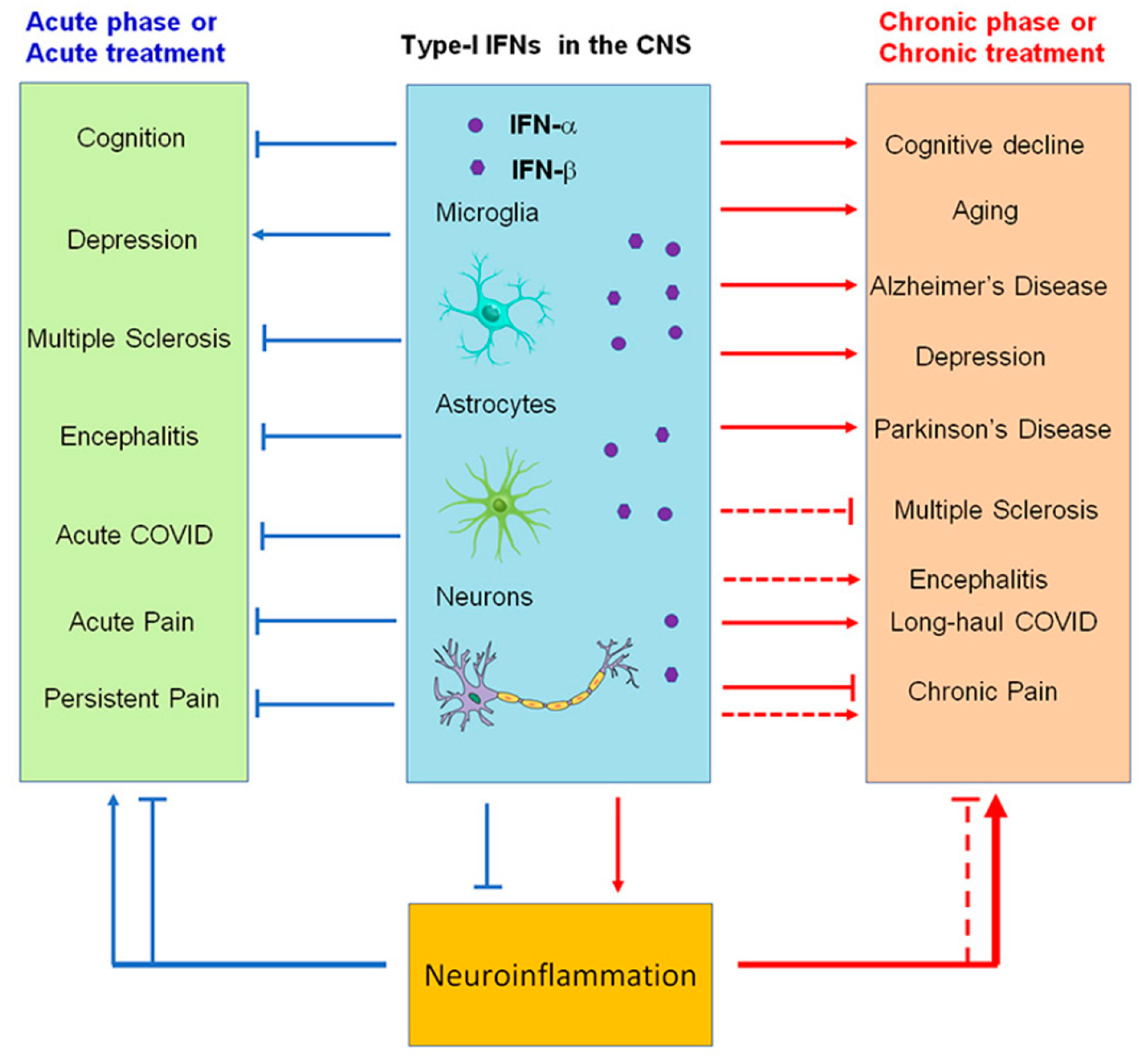
Interferons (IFNs) are pleiotropic cytokines originally identified for their antiviral activity. IFN-α and IFN-β are both type I IFNs that have been used to treat neurological diseases such as multiple sclerosis. Microglia, astrocytes, as well as neurons in the central and peripheral nervous systems, including spinal cord neurons and dorsal root ganglion neurons, express type I IFN receptors (IFNARs). Type I IFNs play an active role in regulating cognition, aging, depression, and neurodegenerative diseases. Notably, by suppressing neuronal activity and synaptic transmission, IFN-α and IFN-β produced potent analgesia.
- neuroinflammation
- neurological disease
- microglia
- astrocytes
- long-haul COVID
- IFN-a
- IFN-b
- spinal cord
1. Introduction
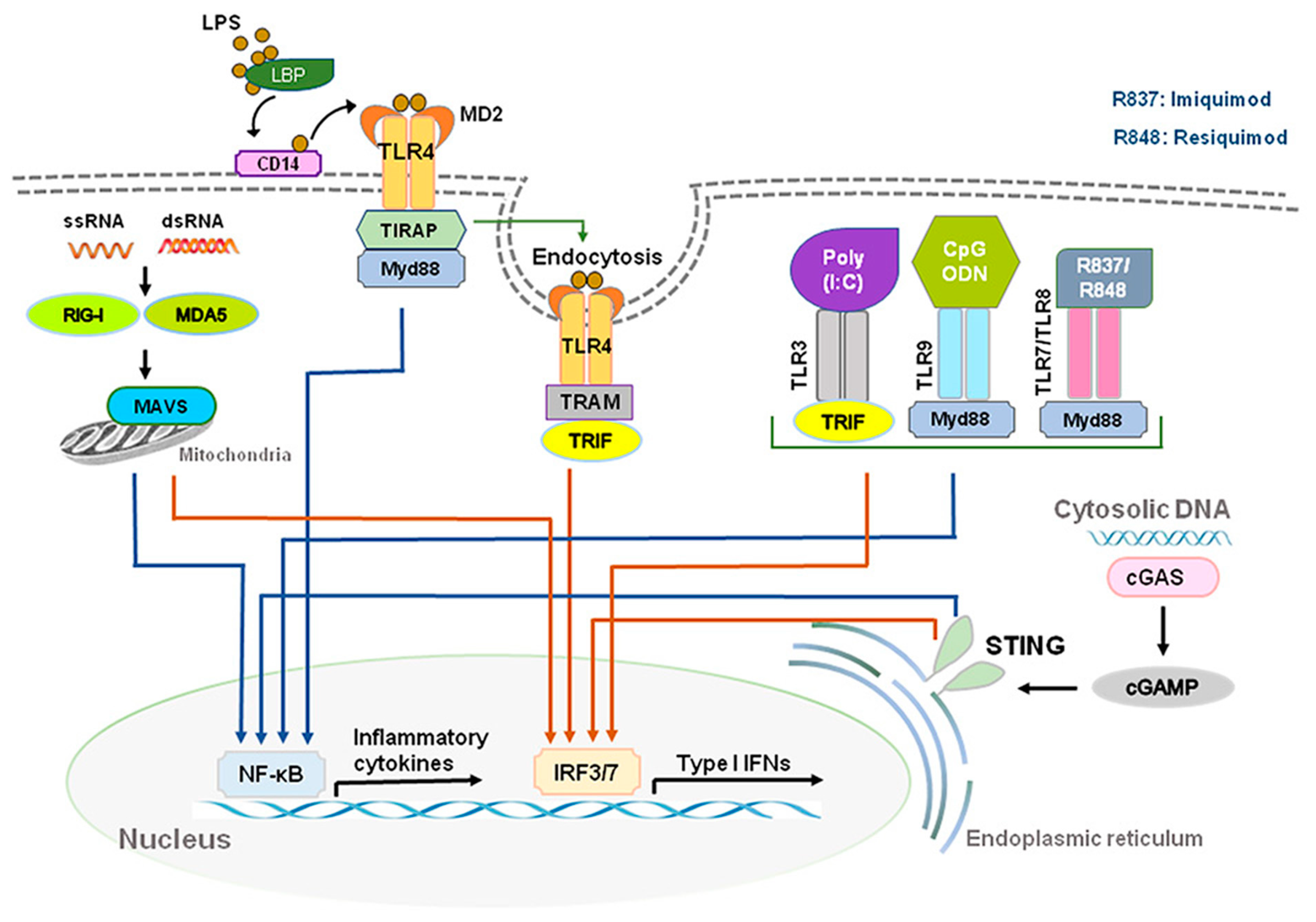
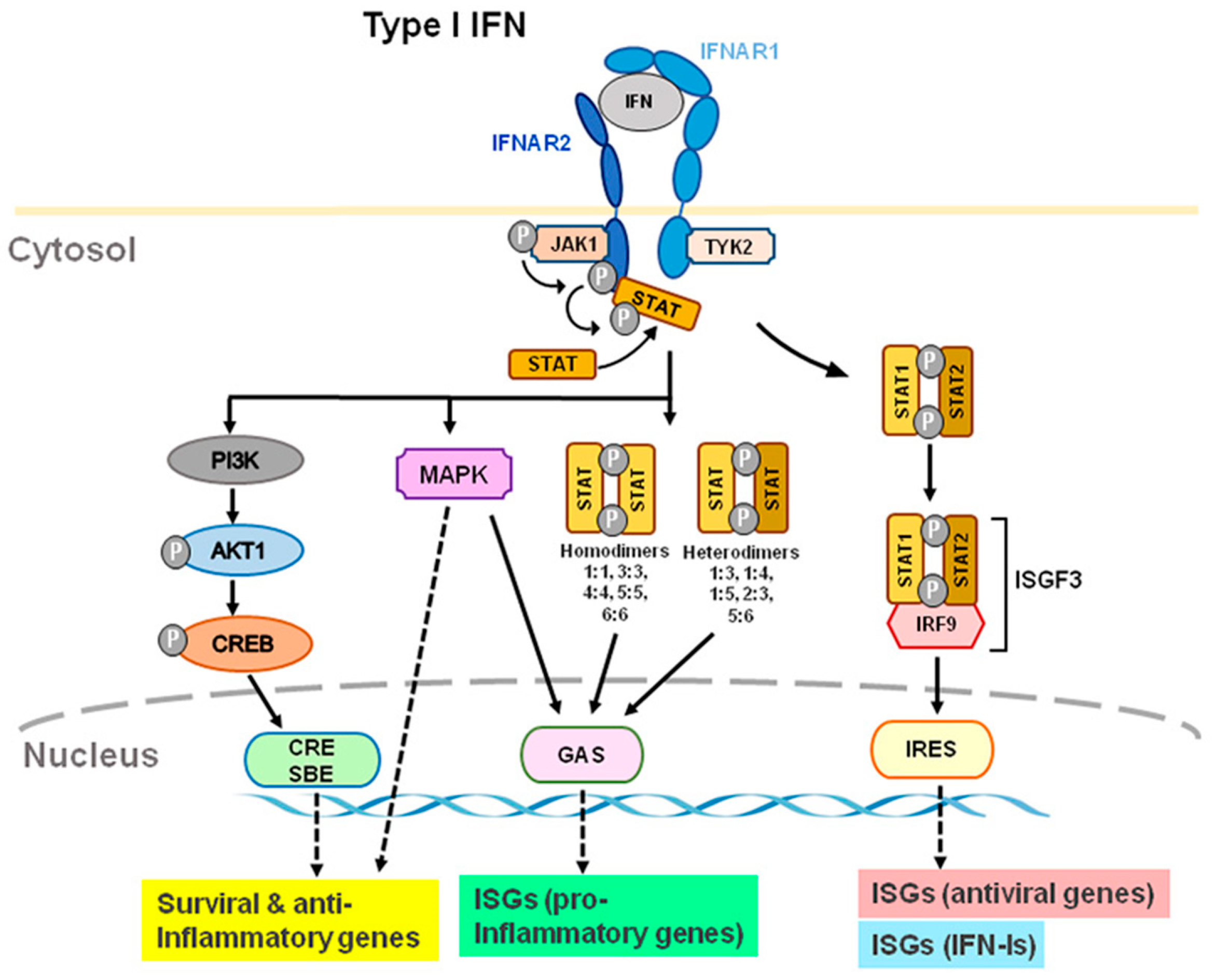
2. Intracellular Signaling of IFN-I
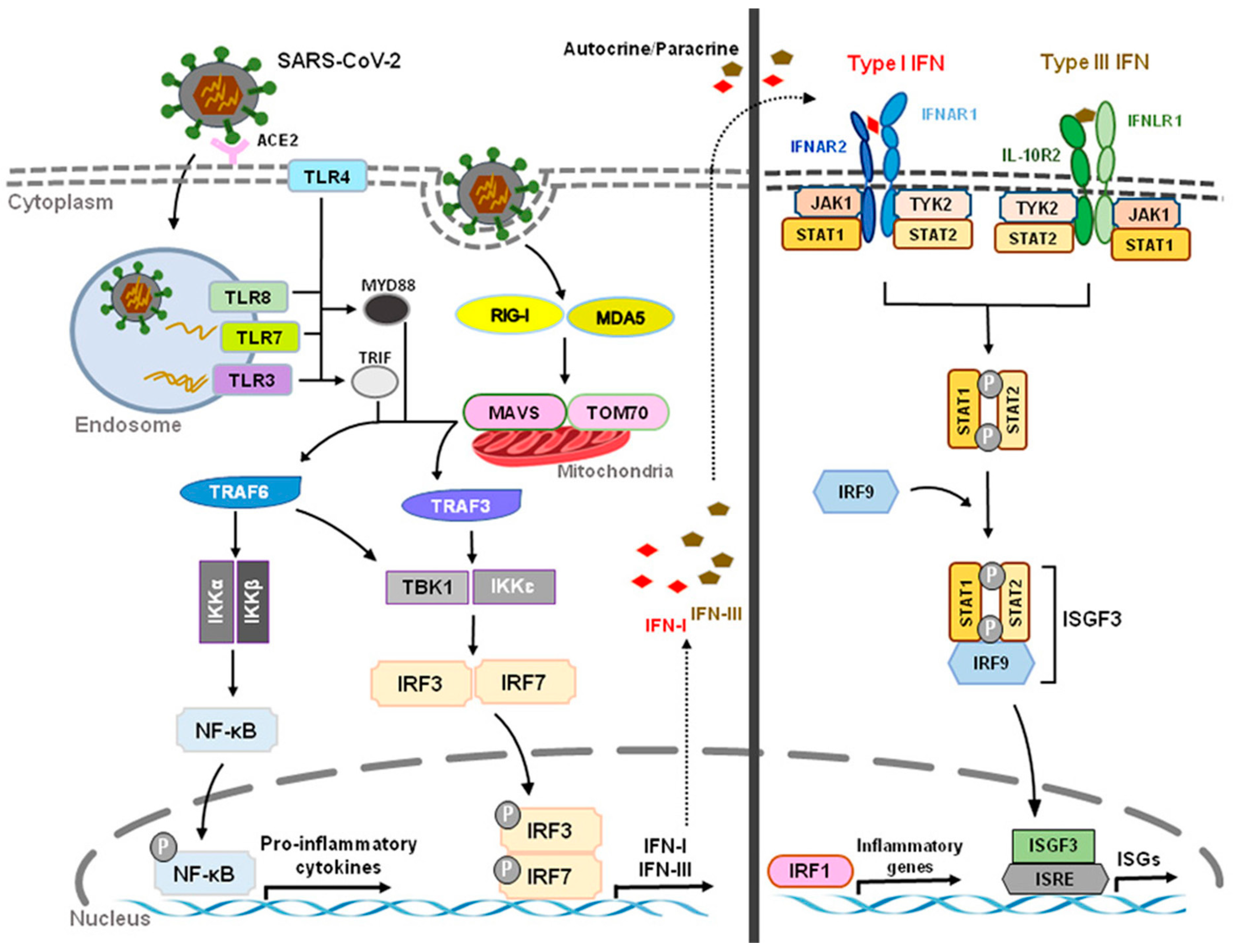
3. Role of IFN-Is in Long-Haul COVID Syndrome
This entry is adapted from the peer-reviewed paper 10.3390/ijms232214394
References
- Pestka, S.; Langer, J.A.; Zoon, K.C.; Samuel, C.E. Interferons and their actions. Annu. Rev. Biochem. 1987, 56, 727–777.
- Goubau, D.; Deddouche, S.; Reis e Sousa, C. Cytosolic sensing of viruses. Immunity 2013, 38, 855–869.
- Pestka, S. The human interferon-alpha species and hybrid proteins. Semin. Oncol. 1997, 24, S9-4–S9-17.
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386.
- Donnelly, C.R.; Jiang, C.; Andriessen, A.S.; Wang, K.; Wang, Z.; Ding, H.; Zhao, J.; Luo, X.; Lee, M.S.; Lei, Y.L.; et al. STING controls nociception via type I interferon signalling in sensory neurons. Nature 2021, 591, 275–280.
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49.
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801.
- Donnelly, C.R.; Chen, O.; Ji, R.R. How Do Sensory Neurons Sense Danger Signals? Trends Neurosci. 2020, 43, 822–838.
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066.
- Liu, T.; Gao, Y.J.; Ji, R.R. Emerging role of Toll-like receptors in the control of pain and itch. Neurosci. Bull. 2012, 28, 131–144.
- Makris, S.; Paulsen, M.; Johansson, C. Type I Interferons as Regulators of Lung Inflammation. Front. Immunol. 2017, 8, 259.
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678.
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792.
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103.
- Asselin-Paturel, C.; Boonstra, A.; Dalod, M.; Durand, I.; Yessaad, N.; Dezutter-Dambuyant, C.; Vicari, A.; O’Garra, A.; Biron, C.; Brière, F.; et al. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat. Immunol. 2001, 2, 1144–1150.
- Biron, C.A. Interferons alpha and beta as immune regulators—A new look. Immunity 2001, 14, 661–664.
- Barchet, W.; Cella, M.; Odermatt, B.; Asselin-Paturel, C.; Colonna, M.; Kalinke, U. Virus-induced interferon alpha production by a dendritic cell subset in the absence of feedback signaling in vivo. J. Exp. Med. 2002, 195, 507–516.
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32.
- Taniguchi, T.; Takaoka, A. A weak signal for strong responses: Interferon-alpha/beta revisited. Nat. Rev. Mol. Cell Biol. 2001, 2, 378–386.
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal. 2017, 15, 23.
- Stanifer, M.L.; Pervolaraki, K.; Boulant, S. Differential Regulation of Type I and Type III Interferon Signaling. Int. J. Mol. Sci. 2019, 20, 1445.
- Chen, J.; Baig, E.; Fish, E.N. Diversity and relatedness among the type I interferons. J. Interferon Cytokine Res. 2004, 24, 687–698.
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421.
- Levin, D.; Schneider, W.M.; Hoffmann, H.H.; Yarden, G.; Busetto, A.G.; Manor, O.; Sharma, N.; Rice, C.M.; Schreiber, G. Multifaceted activities of type I interferon are revealed by a receptor antagonist. Sci. Signal. 2014, 7, ra50.
- Dumitrescu, L.; Constantinescu, C.S.; Tanasescu, R. Recent developments in interferon-based therapies for multiple sclerosis. Expert Opin. Biol. Ther. 2018, 18, 665–680.
- Weise, A.M.; Flaherty, L.E. New options for the adjuvant treatment of cutaneous melanoma? Curr. Oncol. Rep. 2014, 16, 409.
- Snell, L.M.; McGaha, T.L.; Brooks, D.G. Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol. 2017, 38, 542–557.
- Benveniste, E.N.; Qin, H. Type I interferons as anti-inflammatory mediators. Sci. STKE 2007, 2007, pe70.
- Billiau, A. Anti-inflammatory properties of Type I interferons. Antivir. Res. 2006, 71, 108–116.
- Wang, H.; Wang, J.; Xia, Y. Defective Suppressor of Cytokine Signaling 1 Signaling Contributes to the Pathogenesis of Systemic Lupus Erythematosus. Front. Immunol. 2017, 8, 1292.
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9.
- Peluso, M.J.; Kelly, J.D.; Lu, S.; Goldberg, S.A.; Davidson, M.C.; Mathur, S.; Durstenfeld, M.S.; Spinelli, M.A.; Hoh, R.; Tai, V.; et al. Rapid implementation of a cohort for the study of post-acute sequelae of SARS-CoV-2 infection/COVID-19. medRxiv 2021.
- Sudre, C.H.; Keshet, A.; Graham, M.S.; Joshi, A.D.; Shilo, S.; Rossman, H.; Murray, B.; Molteni, E.; Klaser, K.; Canas, L.S.; et al. Anosmia and other SARS-CoV-2 positive test-associated symptoms, across three national, digital surveillance platforms as the COVID-19 pandemic and response unfolded: An observation study. medRxiv 2020.
- Tenforde, M.W.; Billig Rose, E.; Lindsell, C.J.; Shapiro, N.I.; Files, D.C.; Gibbs, K.W.; Prekker, M.E.; Steingrub, J.S.; Smithline, H.A.; Gong, M.N.; et al. Characteristics of Adult Outpatients and Inpatients with COVID-19—11 Academic Medical Centers, United States, March-May 2020. MMWR Morb. Mortal Wkly. Rep. 2020, 69, 841–846.
- Darley, D.R.; Dore, G.J.; Byrne, A.L.; Plit, M.L.; Brew, B.J.; Kelleher, A.; Matthews, G.V. Limited recovery from post-acute sequelae of SARS-CoV-2 at 8 months in a prospective cohort. ERJ Open Res. 2021, 7, 384–2021.
- Lopez-Leon, S.; Wegman-Ostrosky, T.; Perelman, C.; Sepulveda, R.; Rebolledo, P.A.; Cuapio, A.; Villapol, S. More than 50 long-term effects of COVID-19: A systematic review and meta-analysis. Sci. Rep. 2021, 11, 16144.
- Mehandru, S.; Merad, M. Pathological sequelae of long-haul COVID. Nat. Immunol. 2022, 23, 194–202.
- Remsik, J.; Wilcox, J.A.; Babady, N.E.; McMillen, T.A.; Vachha, B.A.; Halpern, N.A.; Dhawan, V.; Rosenblum, M.; Iacobuzio-Donahue, C.A.; Avila, E.K.; et al. Inflammatory Leptomeningeal Cytokines Mediate COVID-19 Neurologic Symptoms in Cancer Patients. Cancer Cell 2021, 39, 276–283.e3.
- Phetsouphanh, C.; Darley, D.R.; Wilson, D.B.; Howe, A.; Munier, C.M.L.; Patel, S.K.; Juno, J.A.; Burrell, L.M.; Kent, S.J.; Dore, G.J.; et al. Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat. Immunol. 2022, 23, 210–216.
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724.
- Channappanavar, R.; Fehr, A.R.; Zheng, J.; Wohlford-Lenane, C.; Abrahante, J.E.; Mack, M.; Sompallae, R.; McCray, P.B., Jr.; Meyerholz, D.K.; Perlman, S. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J. Clin. Investig. 2019, 129, 3625–3639.
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585.
- Chang, S.E.; Feng, A.; Meng, W.; Apostolidis, S.A.; Mack, E.; Artandi, M.; Barman, L.; Bennett, K.; Chakraborty, S.; Chang, I.; et al. New-onset IgG autoantibodies in hospitalized patients with COVID-19. Nat. Commun. 2021, 12, 5417.
- Wang, E.Y.; Mao, T.; Klein, J.; Dai, Y.; Huck, J.D.; Jaycox, J.R.; Liu, F.; Zhou, T.; Israelow, B.; Wong, P.; et al. Diverse functional autoantibodies in patients with COVID-19. Nature 2021, 595, 283–288.
- Zulfiqar, A.A.; Lorenzo-Villalba, N.; Hassler, P.; Andrès, E. Immune Thrombocytopenic Purpura in a Patient with COVID-19. N. Engl. J. Med. 2020, 382, e43.
- Toscano, G.; Palmerini, F.; Ravaglia, S.; Ruiz, L.; Invernizzi, P.; Cuzzoni, M.G.; Franciotta, D.; Baldanti, F.; Daturi, R.; Postorino, P.; et al. Guillain-Barré Syndrome Associated with SARS-CoV-2. N. Engl. J. Med. 2020, 382, 2574–2576.
- Qu, L.; Zhang, P.; LaMotte, R.H.; Ma, C. Neuronal Fc-gamma receptor I mediated excitatory effects of IgG immune complex on rat dorsal root ganglion neurons. Brain Behav. Immun. 2011, 25, 1399–1407.
- Qu, L.; Li, Y.; Pan, X.; Zhang, P.; LaMotte, R.H.; Ma, C. Transient receptor potential canonical 3 (TRPC3) is required for IgG immune complex-induced excitation of the rat dorsal root ganglion neurons. J. Neurosci. 2012, 32, 9554–9562.
- Wang, L.; Jiang, X.; Zheng, Q.; Jeon, S.M.; Chen, T.; Liu, Y.; Kulaga, H.; Reed, R.; Dong, X.; Caterina, M.J.; et al. Neuronal FcγRI mediates acute and chronic joint pain. J. Clin. Investig. 2019, 129, 3754–3769.
- Bersellini Farinotti, A.; Wigerblad, G.; Nascimento, D.; Bas, D.B.; Morado Urbina, C.; Nandakumar, K.S.; Sandor, K.; Xu, B.; Abdelmoaty, S.; Hunt, M.A.; et al. Cartilage-binding antibodies induce pain through immune complex-mediated activation of neurons. J. Exp. Med. 2019, 216, 1904–1924.
- Ferini-Strambi, L.; Salsone, M. COVID-19 and neurological disorders: Are neurodegenerative or neuroimmunological diseases more vulnerable? J. Neurol. 2021, 268, 409–419.
- Heneka, M.T.; Golenbock, D.; Latz, E.; Morgan, D.; Brown, R. Immediate and long-term consequences of COVID-19 infections for the development of neurological disease. Alzheimer’s Res. Ther. 2020, 12, 69.
- Taquet, M.; Luciano, S.; Geddes, J.R.; Harrison, P.J. Bidirectional associations between COVID-19 and psychiatric disorder: Retrospective cohort studies of 62 354 COVID-19 cases in the USA. Lancet Psychiatry 2021, 8, 130–140.
- Naughton, S.X.; Raval, U.; Pasinetti, G.M. Potential Novel Role of COVID-19 in Alzheimer’s Disease and Preventative Mitigation Strategies. J. Alzheimer’s Dis. 2020, 76, 21–25.
- Roy, E.R.; Wang, B.; Wan, Y.W.; Chiu, G.; Cole, A.; Yin, Z.; Propson, N.E.; Xu, Y.; Jankowsky, J.L.; Liu, Z.; et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J. Clin. Investig. 2020, 130, 1912–1930.
- Bauer, K.; Schwarzkopf, L.; Graessel, E.; Holle, R. A claims data-based comparison of comorbidity in individuals with and without dementia. BMC Geriatr. 2014, 14, 10.