The MET gene, known as MET proto-oncogene receptor tyrosine kinase, was first identified to induce tumor cell migration, invasion, and proliferation/survival through canonical RAS-CDC42-PAK-Rho kinase, RAS-MAPK, PI3K-AKT-mTOR, and β-catenin signaling pathways, and its driver mutations, such as MET gene amplification (METamp) and the exon 14 skipping alterations (METex14), activate cell transformation, cancer progression, and worse patient prognosis, principally in lung cancer through the overactivation of their own oncogenic and MET parallel signaling pathways. Because of this, MET driver alterations have become recognized as actionable alterations in lung adenocarcinomas since the FDA approval target therapies for METamp and METex14 in 2020.
- precision medicine
- NSCLC
- target therapies
- resistance mutations
- Driver and actionable mutations
1. Introduction
2. MET in Cancer Initiation and Driver Mutations
2.1. MET Amplification
| Cancer Type | N° GENIE Samples | N° Samples after Filters | METex14 | MET Amp |
|---|---|---|---|---|
| NSCLC | 17,137 | 10,231 | 4% | 2% |
| Renal | 1,986 | 1,556 | 1.2% | 1.2% |
| Hepatobiliary | 2,517 | 1,854 | 0.4% | 1.2% |
| Colorectal | 11,893 | 7,370 | 0% | 0.4% |
| Ovarian | 4,606 | 4,481 | 0.1% | 0.2% |
| Breast | 13,388 | 8,365 | 0% | 0.2% |
| Prostate | 4,379 | 3,530 | 0.1% | 0.1% |
| Total analysis | 56,682 | 36,095 | 5.8% | 5.3% |
2.2. MET Exon 14 Skipping Alterations
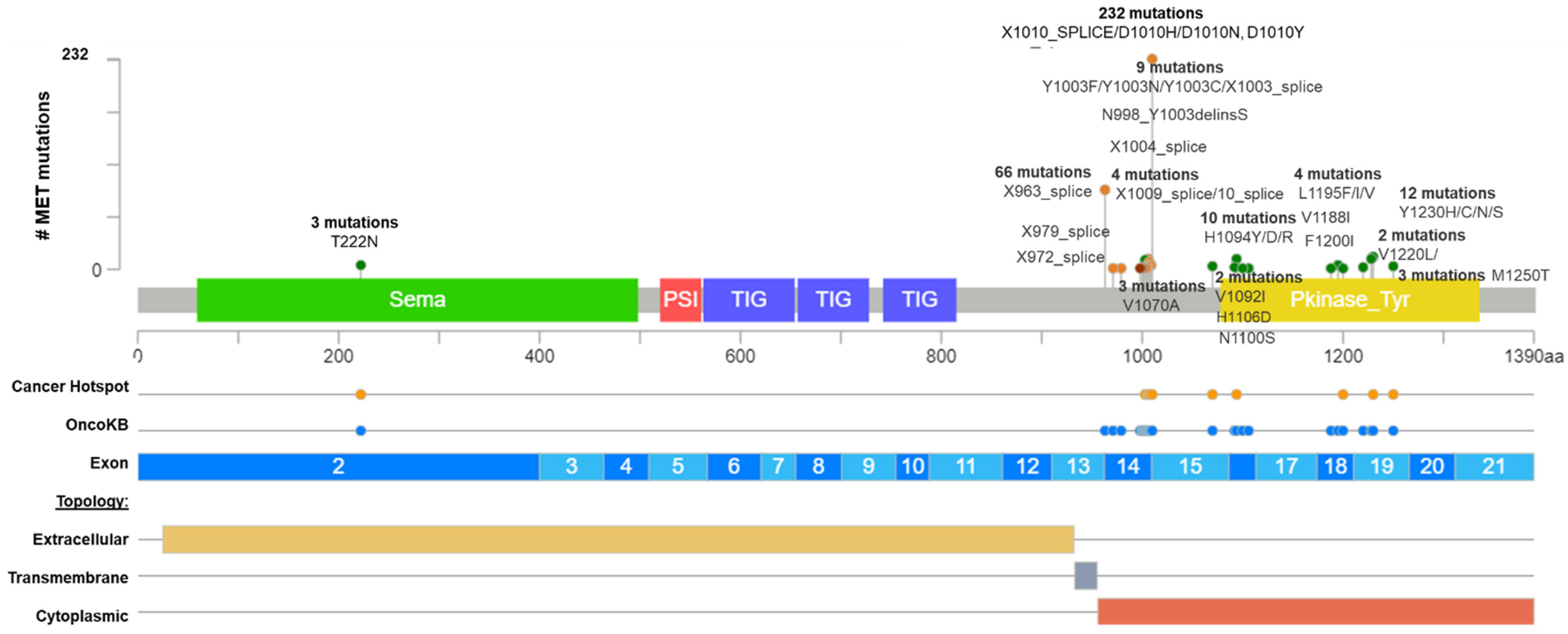
| N° | Protein Change | Variant_ type | exon | CADD13 | SIFT | Mut. Taster | fathm MKL | LRT | M-CAP | MetaLR | Polyphen2 | driver_statement CGI | ONCOGENIC STATEMENT | ONCOKB´S EVIDENCE LEVELS |
| 128 | X1010_splice | Splice_Site | 14 | 25.9 | . | D | D | . | . | . | . | predicted driver: tier 1 | Likely Oncogenic | Level 1 |
| 92 | X963_splice | Splice_Site | 14 | 42 | . | D | D | . | . | . | . | predicted driver: tier 1 | Likely Oncogenic | Level 1 |
| 43 | D1010H | Missense | 14 | 25 | D | D | D | D | D | T | D | predicted driver: tier 1 | Likely Oncogenic | Level 1 |
| 42 | D1010N | Missense | 14 | 25.9 | T | D | D | D | D | T | D | predicted driver: tier 1 | Likely Oncogenic | Level 1 |
| 28 | D1010Y | Missense | 14 | 28.1 | D | D | D | D | D | T | D | predicted driver: tier 1 | Likely Oncogenic | Level 1 |
| 8 | H1094Y | Missense | 16 | 29.0 | D | D | D | D | D | T | D | known in renal_carcinoma | Oncogenic | unknown |
| 8 | X1006_splice | Frame_Shift_Del | 1415 | 27.7 | . | D | D | . | . | . | . | predicted driver: tier 1 | Likely Oncogenic | Level 1 |
| 8 | X1007_splice | Frame_Shift_Del | 1415 | 31 | . | D | D | . | . | . | . | predicted driver: tier 1 | Likely Oncogenic | Level 1 |
| 7 | Y1230H | Missense | 19 | 48 | D | D | D | D | D | D | D | known in renal_carcinoma | Oncogenic | Level R2 |
| 7 | X1008_splice | Frame_Shift_Del | 1415 | 26.7 | . | D | D | . | . | . | . | predicted driver: tier 1 | Likely Oncogenic | Level 1 |
| 5 | D1228N | Missense | 19 | 31 | D | A | D | D | D | D | D | predicted driver: tier 1 | Likely Oncogenic | Level R2 |
| 5 | X1009_splice | In_Frame_ Del | 1415 | 29.6 | . | D | D | . | . | . | . | predicted driver: tier 1 | Likely Oncogenic | Level 1 |
| 4 | D1228H | Missense | 19 | 27.7 | D | D | D | D | D | D | D | predicted driver: tier 1 | likely oncogenic | unknown |
| 4 | T222M | Missense | 2 | 29 | D | D | D | D | D | T | D | predicted passenger | Unknown | unknown |
| 4 | Y1230C | Missense | 19 | 28.7 | D | A | D | D | D | D | D | known in renal_carcinoma | Likely Oncogenic | Level 1 |
| 3 | Y1003N | Missense | 14 | 25.4 | D | D | D | D | D | T | D | predicted driver: tier 1 | Likely oncogenic | unknown |
| 3 | V1070A | Missense | 15 | 32 | D | D | D | D | D | D | D | predicted driver: tier 1 | Likely Oncogenic | unknown |
| 3 | M1250T | Missense | 19 | 31 | D | D | D | D | D | T | D | known in: renal_carcinoma | Oncogenic | Level 1 |
| 3 | Y1230N | Missense | 19 | 26.5 | D | D | D | D | D | D | D | predicted driver: tier 1 | Likely Oncogenic | Level 1 |
| 3 | Y1003F | Missense | 14 | 27.7 | D | D | D | D | D | T | D | predicted driver: tier 1 | Oncogenic | Level 1 |
| 2 | V1220L | Missense | 19 | 27.5 | D | D | D | D | D | T | D | predicted driver: tier 1 | Unknown | unknown |
| 2 | V1092I | Missense | 16 | 31 | D | D | D | D | D | T | D | known in CANCER-PR;carcinoma | Oncogenic | level 1 |
| 2 | D1002G | Missense | 14 | 25.0 | D | D | D | D | D | T | P | predicted driver: tier 1 | Likely Oncogenic | level 1 |
| 2 | Y1003C | Missense | 14 | 28.5 | D | D | D | D | D | T | D | predicted driver: tier 1 | Likely Oncogenic | Level 1 |
| 2 | L1195F | Missense | 18 | 31 | D | D | D | D | D | D | D | predicted driver: tier 1 | Unknown | Level 1 |
| 1 | H1094R | Missense | 16 | 48 | D | A | D | D | D | T | D | known in renal_carcinoma | Oncogenic | unknown |
| 1 | L1195V | Missense | 18 | 27.7 | D | D | D | D | D | D | D | predicted driver: tier 1 | Oncogenic | unknown |
| 1 | V1220I | Missense | 19 | 28.2 | D | A | D | D | D | T | D | predicted driver: tier 1 | Likely Oncogenic | unknown |
| 1 | Y1230S | Missense | 19 | 26.8 | D | D | D | D | D | D | D | predicted driver: tier 1 | Likely Oncogenic | unknown |
| 1 | N998_Y1003delinsS | In_Frame_ Del | 14 | 48 | . | D | D | . | . | . | . | predicted driver: tier 1 | Unknown | unknown |
| 1 | H1094D | Missense | 16 | 26.8 | D | D | D | D | D | D | D | predicted driver: tier 1 | Unknown | unknown |
| 1 | V1188I | Missense | 18 | 29.1 | D | D | D | D | D | T | D | predicted driver: tier 1 | Unknown | unknown |
| 1 | L1195I | Missense | 18 | 48 | D | D | D | D | D | D | D | predicted driver: tier 1 | unknown | unknown |
| 1 | X972_splice | Frame_Shift_Del | 1415 | 24.9 | . | D | D | . | . | . | . | predicted passenger | unknown | unknown |
| 1 | X979_splice | Frame_Shift_Del | 1415 | 29.3 | . | D | D | . | . | . | . | predicted passenger | unknown | unknown |
| 1 | X1001_splice | In_Frame_Del | 1415 | 48 | . | D | D | . | . | . | . | predicted passenger | unknown | unknown |
| 1 | X1003_splice | Frame_Shift_Del | 1415 | 48 | . | D | D | . | . | . | . | predicted passenger | unknown | unknown |
| 1 | X1004_splice | Frame_Shift_Del | 1415 | 24.9 | . | D | D | . | . | . | . | predicted passenger | unknown | unknown |
| 1 | T1006_D1010delinsN | In_Frame_ Del | 14 | 48 | . | D | D | . | . | . | . | predicted passenger | unknown | unknown |
| 1 | F1007_D1010delinsY | In_Frame_ Del | 14 | 48 | . | D | D | . | . | . | . | predicted driver: tier 1 | likely oncogenic | unknown |
| 1 | N1100S | Missense | 16 | 48 | T | N | N | N | T | T | B | predicted passenger | unknown | unknown |
| 1 | D1228V | Missense | 19 | 26.7 | D | D | D | D | D | D | D | known in LUAD | Likely oncogenic | unknown |
| 1 | H1106D | Missense | 16 | 48 | D | D | D | D | D | T | D | predicted driver: tier 1 | Likely Oncogenic | Level R2 |
| 1 | F1200I | Missense | 18 | 32 | D | D | D | D | D | T | D | predicted driver: tier 1 | Oncogenic | Level R2 |
| 1 | V1001_F1007del | In_Frame_Del | 14 | 25.9 | . | D | D | . | . | . | . | predicted driver: tier 1 | Likely Oncogenic | Level 1 |
| 1 | D1002_F1007del | In_Frame_Del | 14 | 41 | . | D | D | . | . | . | . | predicted driver: tier 1 | Likely Oncogenic | Level 1 |
This entry is adapted from the peer-reviewed paper 10.3390/ijms232213898
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Cooper, C.S.; Park, M.; Blair, D.G.; Tainsky, M.A.; Huebner, K.; Croce, C.M.; Vande Woude, G.F. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 1984, 311, 29–33.
- Wang, R.; Ferrell, L.D.; Faouzi, S.; Maher, J.J.; Bishop, J.M. Activation of the Met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice. J. Cell Biol. 2001, 153, 1023–1034.
- Tward, A.D.; Jones, K.D.; Yant, S.; Cheung, S.T.; Fan, S.T.; Chen, X.; Kay, M.A.; Wang, R.; Bishop, J.M. Distinct pathways of genomic progression to benign and malignant tumors of the liver. Proc. Natl. Acad. Sci. USA 2007, 104, 14771–14776.
- Mi, J.; Hooker, E.; Balog, S.; Zeng, H.; Johnson, D.T.; He, Y.; Yu, E.J.; Wu, H.; Le, V.; Lee, D.H.; et al. Activation of hepatocyte growth factor/MET signaling initiates oncogenic transformation and enhances tumor aggressiveness in the murine prostate. J. Biol. Chem. 2018, 293, 20123–20136.
- Ichimura, E.; Maeshima, A.; Nakajima, T.; Nakamura, T. Expression of c-met/HGF receptor in human non-small cell lung carcinomas in vitro and in vivo and its prognostic significance. Jpn. J. Cancer Res. 1996, 87, 1063–1069.
- Siegfried, J.M.; Weissfeld, L.A.; Luketich, J.D.; Weyant, R.J.; Gubish, C.T.; Landreneau, R.J. The clinical significance of hepatocyte growth factor for non-small cell lung cancer. Ann. Thorac. Surg. 1998, 66, 1915–1918.
- Overbeck, T.R.; Cron, D.A.; Schmitz, K.; Rittmeyer, A.; Korber, W.; Hugo, S.; Schnalke, J.; Lukat, L.; Hugo, T.; Hinterthaner, M.; et al. Top-level MET gene copy number gain defines a subtype of poorly differentiated pulmonary adenocarcinomas with poor prognosis. Transl. Lung Cancer Res. 2020, 9, 603–616.
- Yin, W.; Cheng, J.; Tang, Z.; Toruner, G.; Hu, S.; Guo, M.; Robinson, M.; Medeiros, L.J.; Tang, G. MET Amplification (MET/CEP7 Ratio >/= 1.8) Is an Independent Poor Prognostic Marker in Patients With Treatment-naive Non-Small-cell Lung Cancer. Clin. Lung Cancer 2021, 22, e512–e518.
- Kim, J.H.; Kim, H.S.; Kim, B.J.; Jang, H.J.; Lee, J. Prognostic value of c-Met overexpression in hepatocellular carcinoma: A meta-analysis and review. Oncotarget 2017, 8, 90351–90357.
- Yang, Y.; Wang, C.; Dai, C.; Liu, X.; Li, W.; Huang, M.; Zhao, X.; Ji, D.; Li, J.; Guo, W. Amplification and expression of c-MET correlate with poor prognosis of patients with gastric cancer and upregulate the expression of PDL1. Acta Biochim. Biophys. Sin. (Shanghai) 2021, 53, 547–557.
- Zagouri, F.; Bago-Horvath, Z.; Rossler, F.; Brandstetter, A.; Bartsch, R.; Papadimitriou, C.A.; Dimitrakakis, C.; Tsigginou, A.; Papaspyrou, I.; Giannos, A.; et al. High MET expression is an adverse prognostic factor in patients with triple-negative breast cancer. Br. J. Cancer 2013, 108, 1100–1105.
- Noonan, S.A.; Berry, L.; Lu, X.; Gao, D.; Baron, A.E.; Chesnut, P.; Sheren, J.; Aisner, D.L.; Merrick, D.; Doebele, R.C.; et al. Identifying the Appropriate FISH Criteria for Defining MET Copy Number-Driven Lung Adenocarcinoma through Oncogene Overlap Analysis. J. Thorac. Oncol. 2016, 11, 1293–1304.
- Consortium, A.P.G. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831.
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404.
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1.
- Frampton, G.M.; Ali, S.M.; Rosenzweig, M.; Chmielecki, J.; Lu, X.; Bauer, T.M.; Akimov, M.; Bufill, J.A.; Lee, C.; Jentz, D.; et al. Activation of MET via Diverse Exon 14 Splicing Alterations Occurs in Multiple Tumor Types and Confers Clinical Sensitivity to MET Inhibitors. Cancer Discov. 2015, 5, 850–859.
- Paik, P.K.; Drilon, A.; Fan, P.D.; Yu, H.; Rekhtman, N.; Ginsberg, M.S.; Borsu, L.; Schultz, N.; Berger, M.F.; Rudin, C.M.; et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015, 5, 842–849.
- Onozato, R.; Kosaka, T.; Kuwano, H.; Sekido, Y.; Yatabe, Y.; Mitsudomi, T. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J. Thorac. Oncol. 2009, 4, 5–11.
- Kong-Beltran, M.; Seshagiri, S.; Zha, J.; Zhu, W.; Bhawe, K.; Mendoza, N.; Holcomb, T.; Pujara, K.; Stinson, J.; Fu, L.; et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006, 66, 283–289.
- Kron, A.; Scheffler, M.; Heydt, C.; Ruge, L.; Schaepers, C.; Eisert, A.K.; Merkelbach-Bruse, S.; Riedel, R.; Nogova, L.; Fischer, R.N.; et al. Genetic Heterogeneity of MET-Aberrant NSCLC and Its Impact on the Outcome of Immunotherapy. J. Thorac. Oncol. 2021, 16, 572–582.
- Sattler, M.; Salgia, R. MET in the Driver’s Seat: Exon 14 Skipping Mutations as Actionable Targets in Lung Cancer. J. Thorac. Oncol. 2016, 11, 1381–1383.
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017.
- Subramanian, J.; Tawfik, O. Detection of MET exon 14 skipping mutations in non-small cell lung cancer: Overview and community perspective. Expert Rev. Anticancer Ther. 2021, 21, 877–886.
- Socinski, M.A.; Pennell, N.A.; Davies, K.D. MET Exon 14 Skipping Mutations in Non-Small-Cell Lung Cancer: An Overview of Biology, Clinical Outcomes, and Testing Considerations. JCO Precis. Oncol. 2021, 5.
- Awad, M.M.; Lee, J.K.; Madison, R.; Classon, A.; Kmak, J.; Frampton, G.M.; Alexander, B.M.; Venstrom, J.; Schrock, A.B. Characterization of 1,387 NSCLCs with MET exon 14 (METex14) skipping alterations (SA) and potential acquired resistance (AR) mechanisms. J. Clin. Oncol. 2020, 38, 9511.
- Poirot, B.; Doucet, L.; Benhenda, S.; Champ, J.; Meignin, V.; Lehmann-Che, J. MET Exon 14 Alterations and New Resistance Mutations to Tyrosine Kinase Inhibitors: Risk of Inadequate Detection with Current Amplicon-Based NGS Panels. J. Thorac. Oncol. 2017, 12, 1582–1587.
- Davies, K.D.; Lomboy, A.; Lawrence, C.A.; Yourshaw, M.; Bocsi, G.T.; Camidge, D.R.; Aisner, D.L. DNA-Based versus RNA-Based Detection of MET Exon 14 Skipping Events in Lung Cancer. J. Thorac. Oncol. 2019, 14, 737–741.
- Kim, E.K.; Kim, K.A.; Lee, C.Y.; Kim, S.; Chang, S.; Cho, B.C.; Shim, H.S. Molecular Diagnostic Assays and Clinicopathologic Implications of MET Exon 14 Skipping Mutation in Non-small-cell Lung Cancer. Clin. Lung Cancer 2019, 20, e123–e132.