1. EGFR-TKIs
The ErbB family of RTKs, which also includes ErbB-1 (HER1, EGFR), ErbB-2 (HER2, Neu), ErbB-3 (HER3), and ErbB-4, comprises the transmembrane glycoprotein known as EGFR (HER4). When EGFR binds to ligands, specific intracellular signaling pathways, including PI3K/Akt and MAPK, which are involved in the proliferation, differentiation, migration, and death of some cells, are stimulated (
Figure 1) [
10].
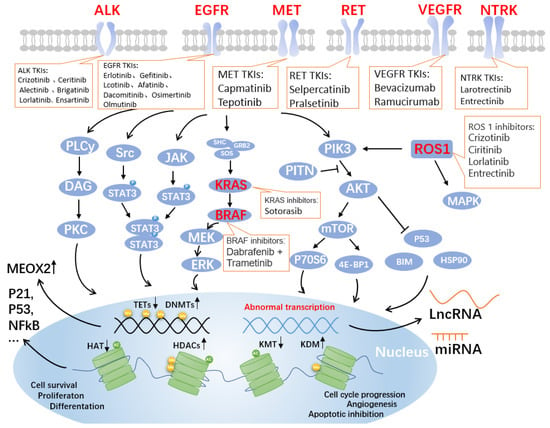
Figure 1. Genes and pathways associated with targeted drugs for NSCLC. Four critical signaling pathways include JAK-STAT, MAPK, PLC-gamma (phospho-lipase C gamma), and PI3K-AKT. These pathways are well-known controllers of cell cycle progression, proliferation, and apoptosis/cell survival; deregulation is a frequent characteristic of human malignancies. Alterations in key pathways will affect DNA methylation modifications, such as increased DNA methyltransferases (DNMTs) and decreased the ten-eleven translocation methylcytosine dioxygenases (TETs), further allowing overexpression of mesenchymal homology box 2 (MEOX2), whose expression is negatively correlated with patient survival. Additionally, post-translational histone modifications were affected. As shown in the figure, histone acetyltransferases (HATs), histone deacetylases (HDACs, also known as lysine deacetylases or KDACs), the lysine methyltransferases (KMTs) and lysine demethylases (KDMs) undergo corresponding up- or downregulation, affecting the expression of P21, P53, nuclear factor κB (NFκB), and other related proteins that are closely related to the cell cycle. Non-coding RNAs, such as long non-coding RNAs (LncRNAs) and miRNAs, are produced as a result of abnormal transcription. The lncRNA is a brand-new class of regulatory RNA. The LncRNA HOX antisense intergenic RNA (HOTAIR), an oncogene in NSCLC, is one of the significant factors controlling the growth of malignancies. Unknown are the immunomodulatory pathway and probable molecular mechanism involved in NSCLC. Notably, the graphic labels current FDA-approved medications that target EGFR, ALK, MET, RET, VEGF, NTRK, ROS1, KRAS, and BRAF.
Since EGFR is expressed by more than 60% of NSCLCs, it has become a crucial therapeutic target for treating these malignancies. Inhibitors targeting the structural domain of tyrosine kinase inhibitors (TKIs) have been developed and are clinically active. Moreover, these TKIs are especially effective in patients who contain activating mutations in the tyrosine kinase structural domain of the EGFR gene [
11].
Erlotinib and gefitinib are examples of first-generation medications that are reversible inhibitors. Erlotinib’s group was shown to have a median progression-free survival (PFS) of 9.7 months, while the group receiving conventional chemotherapy had a median PFS of 5.2 months [
12]. Afatinib and dacomitinib are examples of irreversible second-generation inhibitors that bind to EGFR covalently. In contrast to platinum-based chemotherapy, patients with EGFR-mutant cancers showed >70% radiological response times and statistically significantly improved PFS when treated with first-generation (erlotinib and gefitinib) or second-generation (afatinib) EGFR TKIs (
Table 1) [
10,
13,
14,
15].
For metastatic EGFR-mutant NSCLC patients who have developed the EGFR T790M resistance mutation, osimertinib was the first third-generation EGFR TKI to obtain FDA and EMA approval [
16]. For patients with EGFR-mutant NSCLC, osimertinib is superior to erlotinib and gefitinib as the first-line treatment [
17].
2. ALK-TKIs
The ALK gene encodes a tyrosine kinase receptor and is located on the short arm of chromosome 2 (2p23), belongs to the insulin receptor superfamily, and encodes for the ALK protein. The oncogenic ALK fusion gene is present in 3–5% of NSCLC patients [
22]. ALK is a transmembrane tyrosine kinase receptor that functions similarly to other RTKs in that it has an extracellular domain, a membrane segment, and a cytoplasmic receptor kinase region [
23,
24]. In NSCLC, more than 19 distinct ALK fusion partners, including EML4, KIF5B, KLC1, and TPR, have been identified [
25]. About 85% of all fusion variants in ALK+ NSCLC are represented by the prevalent fusion variant, EML4-ALK. Additionally, the most frequent genetic co-alterations in ALK+ NSCLC are TP53 mutations [
25].
The first oral ALK TKI approved for the treatment of non-small cell lung cancer (NSCLC) that was positive for ALK, crizotinib, initially showed promising outcomes (
Table 2). The initial euphoria, however, was subdued because almost all of the treated individuals unavoidably developed resistance within a year and experienced disease progression, mainly in the brain or other parenchymal areas [
26]. When crizotinib binds to the ATP pocket of the MET kinase in a DFG-in conformation, it forms conventional hydrogen bonds (Hb) with the residues in the hinge area. Additionally, the activation loop and its phenyl ring interact poorly (A-loop). The medication was discovered to have unintended effects on ALK and other kinases [
8]. Second-generation ALK TKIs such as crizotinib (LDK378), alectinib (CH5424802/RO5424802), and brigatinib (AP26113) were developed to combat therapy-induced acquired resistance and boost efficacy in ALK-positive patients receiving crizotinib pretreatment, even those with metastases to the central nervous system (CNS) (
Table 2). [
27,
28]. Alectinib forms a classical Hb with M1199 by attaching to the ATP-binding site of ALK. Additionally, alectinib interacts with several additional nearby residues from the -helix (K1150, E1167), the catalytic loop (R1253), and the DFG motif via solvent water molecules (G1269, D1270). As a result, the substance is a part of a stabilizing global Hb network that may likely make up for any one mutation at the binding site [
8,
29]. Furthermore, third-generation ALK TKIs, including lorlatinib (PF-06463922), entrectinib (RxDx-101), and ensartinib (X-398), provided promising early findings in terms of both clinical activity and safety, according to recent clinical trials (
Table 2). [
30].
3. Other Targeted Sites and Drugs for NSCLC
3.1. ROS1
The ROS proto-oncogene 1 is a member of the insulin receptor subfamily and is encoded by the ROS1 gene on chromosome 6Q22.1 [
35]. It has a sizable hydrophobic single-pass transmembrane region, an extensive N-terminal extracellular structural domain, and a C-terminal intracellular tyrosine kinase structural domain [
36]. ROS1 rearrangements, a fusion that encourages tumorigenicity and/or independent growth of different cell lines, are present in 1–2% of NSCLC patients [
37,
38], and these patients are more likely to be female and to have smoked less [
39]. The median age of the 29 individuals with ROS1 rearrangement was 51, ranging from 30 to 80 years old, and 68.9% of them had never smoked [
40]. At first glance, the proportion may seem small, but given the massive base of NSCLC patients, it is estimated that there are 10,000–15,000 new cases of the disease worldwide each year [
35].
Phylogenetic sequence analysis identified that ROS1 has been linked to the ALK/LTK and insulin receptor RTK families. Homology with ALK is very significant in the development of ROS1-directed medications; nevertheless, not all ALK TKI exhibit dual inhibitory activity against ALK and ROS1. In 2016, the U.S. FDA and the European Medicines Agency approved the drug crizotinib, a multitargeted MET, ALK, and ROS1 inhibitor that showed considerable efficacy in NSCLCs with ROS1 rearrangements in a phase I study [
41]. The ROS1 expansion group of crizotinib’s phase I trial had an objective response rate (ORR) of 72%. The overall response duration was 17.6 months, whereas the median PFS was 19.2 months [
42]. Four drugs with notable action against ROS1+ NSCLC are FDA-approved: crizotinib, ciritinib, lorlatinib, and entrectinib (
Table 3). Entrectinib, lorlatinib, and ciritinib all had an overall response rate of more than 60%, with entrectinib having an intracranial activity [
43].
3.2. BRAF
The serine/threonine protein kinase family includes mutations in the v-RAF murine sarcoma viral oncogene homolog B (BRAF), a crucial effector molecule for the MAPK/ERK signaling pathway (
Figure 1). BRAF mutations are present in 4% of NSCLC, and 50% of these mutations are not V600 variants [
44]. By breaking the glycine-rich P loop and its variant domain of the kinase segment, somatic mutations in BRAF that result in the V600E variation change two major areas of the peptide. In WT BRAF, the transition between the active and inactive states is accomplished by activating the inhibitory effect caused by the glycine-rich P loop, which is crucial for incorporating the signal transduction supplied by RAS [
45,
46]. There is no preference for race in the prevalence of BRAF mutant lung cancer, which ranges from 1.5% to 3.5% [
47].
The FDA expanded the use of dabrafenib and trametinib on 22 June 2017, allowing for the treatment of patients with metastatic NSCLC who have the BRAF(V600E) mutation [
48]. A two-cohort phase II study compared patients treated with dabrafenib as a single agent with dabrafenib in combination with trametinib and found that the ORR was 33% vs. 67% and the PFS was 5.5 vs. 10.2 months, respectively [
49]. Additionally, the French National Cancer Institute (INCA) experiment showed that BRAF(V600E) mutation-positive NSCLC patients responded well to vemurafenib monotherapy, although BRAF(nonV600) mutation-positive individuals did not [
50]. For patients with advanced or metastatic melanoma, non-small cell lung cancer, or anaplastic thyroid cancer and BRAF(V600E/K) mutations, the U.S. FDA has currently approved three RAF and MEK inhibitor combinations: vemurafenib/cobimetinib (Genentech, San Francisco, CA, USA), dabrafenib/trametinib (Novartis, Basel, Switzerland), and encorafenib/binimetinib (Array BioPharma, Boulder, CO, USA) [
51].
3.3. MET
The MET receptor is located on the long arm of human chromosome 7 (7q31) and is encoded by the MET oncogene. This oncogene was first identified in a human osteosarcoma cell line containing the transforming fusion protein TPR–MET, generated by a rearrangement between a translocation promoter region (TPR) located on chromosome 1 at the 5’ end and the MET gene located on chromosome 7 at the 3’ end [
52,
53]. HGF ligand binding to the MET receptor causes homodimerization and phosphorylation of intracellular tyrosine residues, which activates MET [
54]. This triggers the downstream signaling pathways for RAS/ERK/MAPK, PI3K-AKT, Wnt/catenin, and STAT [
55].
Small cell lung cancer was the first disease to be linked to somatic mutations affecting splicing sites of exon 14 of the MET gene, which codes for the juxtamembrane region [
56]. The median age was 61 years for patients with EGFR mutations, 65 years for KRAS mutant NSCLC, and a significantly older median age of 72.5 years for patients with MET exon 14 mutant NSCLC. Overall, 36% of MET exon 14 mutation patients had never smoked, and 68% were female [
57]. At least seven TKIs targeting MET gene mutations are currently on the market or in clinical trials, including crizotinib, cabozantinib, voritinib, tepotinib, capmatinib, glesatinib, and merestinib, with additional drugs in preclinical studies [
58]. Tepotinib, capmatinib, and savolitinib have all demonstrated potent actions in phase I/II investigations; in fact, tepotinib and capmatinib were approved for usage by health authorities [
59]. Tepotinib and capmatinib received FDA approval on 3 February 2021, and 6 May 2020, respectively. Patients with metastatic non-small cell lung cancer (mNSCLC) whose tumors carry an exon 14 skipping mutation associated with the mesenchymal–epithelial transition (MET) are advised to take capmatinib. Tepotinib is recommended for people with mNSCLC who had MET exon 14 skipping mutations [
60].
3.4. RET
Transmembrane glycoprotein receptor-tyrosine kinase is produced during transfection by the RET (rearranged during transfection) proto-oncogene, which is located on chromosome 10 [
61]. The RET gene can be found in 1% to 2% of all NSCLC patients undergoing chromosomal rearrangement and is involved in various upstream fusion partners, such as KIF5B, TRIM33, CCDC6, and NCOA4 [
62]. Multitarget inhibitors with anti-rearranged during RET action have been studied in patients with RET-rearranged lung cancer in several preclinical models, clinical trials, and retrospective investigations to date. The advantage in terms of response (16–47%) and PFS (2–7 months) in the clinical situation is not comparable to that reported with other targeted medicines in NSCLC patients with oncogene addiction [
63]. The FDA approved pralsetinib in September 2020 for the treatment of people with metastatic RET fusion-positive NSCLC [
64]. This is the first oral tyrosine kinase inhibitor that can be taken once a day by people with metastatic NSCLC that is RET fusion positive. Patients who had received platinum-based chemotherapy in the past or had just started treatment were shown to have response rates of 57% and 70%, respectively, to pralsetinib [
65].
3.5. KRAS
The proto-oncogene KRAS (Kirsten rat sarcoma 2 viral oncogene homolog) produces the small GTPase transductor protein KRAS [
66]. Overall, KRAS accounts for 85% of RAS mutations observed in human cancers, and KRAS(G12C) mutation occurs in 13% of NSCLCs [
67]. Although the panorama of treatment for advanced NSCLC has been significantly altered in recent years by the use of targeted therapies and immune checkpoint inhibitors, past attempts to target KRAS (direct and indirect approaches) have not been particularly successful [
67]. The RAF-MEK-ERK pathway is one of the cell growth and division pathways that is promoted by KRAS(G12C) mutations [
68]. For the treatment of adult patients with locally advanced or metastatic NSCLC with KRAS (G12C) mutations who have undergone at least one prior systemic therapy as established by the FDA-approved test, sotorasib was given accelerated approval by the FDA in May 2021 [
69]. In a phase I study, sotorasib demonstrated antitumor effects in patients with advanced solid tumors bearing the KRAS (G12C) mutation. In a single-arm phase II trial, 33.9% of patients had partial remissions and 4.2% had complete remissions, making up the total number of patients who had objective remissions. The average length of remission was 11.1 months [
70,
71]. Sotorasib, an oral small molecule inhibitor of the RAS GTPase family, irreversibly binds to the P2 pocket of inactive GDP-bound KRAS. The cysteine in KRAS (G12C) establishes an irreversible covalent bond with sotorasib, immobilizing the protein in an inactive state. By preventing KRAS signaling, sotorasib inhibits both in vitro and in vivo cell growth as well as tumor growth, and it only causes apoptosis in KRAS (G12C) tumor cell lines [
68,
72].
3.6. VEGF
The growth factor known as vascular endothelial growth factor (VEGF) has significant pro-angiogenic activity and affects endothelial cells in a mitogenic and anti-apoptotic manner. It also enhances vascular permeability and encourages cell migration. These results mean that it actively contributes to the regulation of both healthy and unhealthy angiogenic processes [
73].
The first VEGF inhibitor to be authorized for cancer treatment is bevacizumab. The U.S. FDA, the European Medicines Agency (EMEA), and numerous other regulatory bodies have approved bevacizumab for treating malignancies such as NSCLC at this time [
73]. Bevacizumab or ramucirumab added to EGFR TKIs significantly increased PFS in patients with EGFR-mutant NSCLC in recently published large extensive randomized studies [
74], In a phase III trial, the inclusion of bevacizumab significantly increased the PFS endpoint from 11.2 months when erlotinib was used alone to 17.9 months when it was used in combination therapy [
75].
This entry is adapted from the peer-reviewed paper 10.3390/ijms232315056