Type 1 diabetes (T1D) is a multifactorial autoimmune disease driven by T-cells against the insulin-producing islet β-cells, resulting in a marked loss of β-cell mass and function. A dysbiotic gut microbial profile has been associated with T1D patients. Moreover, new evidence propose that perturbation in gut microbiota may influence the T1D onset and progression. One of the prominent features in clinically silent phase before the onset of T1D is the presence of a microbiota characterized by low numbers of commensals butyrate producers, thus negatively influencing the gut permeability. The loss of gut permeability leads to the translocation of microbes and microbial metabolites and could lead to the activation of immune cells. Moreover, microbiota-based therapies to slow down disease progression or reverse T1D have shown promising results.
- type 1 diabetes (T1D)
- insulin resistance
- gut microbiome
- dysbiosis
1. Introduction
2. T1D Risk Factors
3. Role of Gut Microbiota in T1D Pathophysiology
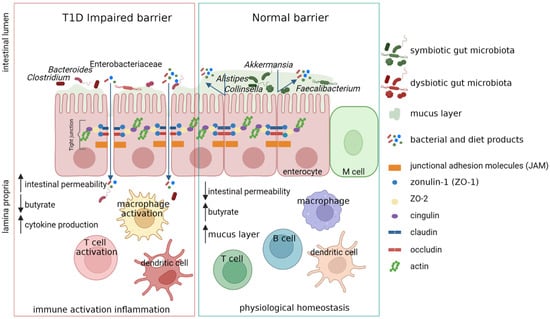
4. Gut Microbiota Dysbiosis in T1D
5. Conclusion
Although T1D was earlier regarded to have genetic roots, compelling evidences state a strong role of Gut microbiota in disease onset and progression. Thus, the correction of dysbiosis by microbial-based therapies could help in promoting immune tolerance at onset, and improve gut permeability decreasing inflammation during the disease progression.
This entry is adapted from the peer-reviewed paper 10.3390/ijms232314650
References
- Maahs, D.M.; West, N.A.; Lawrence, J.M.; Mayer-Davis, E.J. Epidemiology of Type 1 Diabetes. Endocrinol. Metab. Clin. N. Am. 2010, 39, 481–497.
- Karuranga, S.; Malanda, B.; Saeedi, P.; Salpea, P. (Eds.) IDF Diabetes Atlas, 9th Edition Committee IDF DIABETES ATLAS Ninth Edition 2019; IDF: Brussels, Belgium, 2019; ISBN 978-2-930229-87-4.
- Lucier, J.; Weinstock, R.S. Diabetes Mellitus Type 1. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022.
- Taplin, C.E.; Barker, J.M. Autoantibodies in Type 1 Diabetes. Autoimmunity 2008, 41, 11–18.
- Insel, R.A.; Dunne, J.L.; Atkinson, M.A.; Chiang, J.L.; Dabelea, D.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Krischer, J.P.; Lernmark, Å.; et al. Staging Presymptomatic Type 1 Diabetes: A Scientific Statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015, 38, 1964–1974.
- MetaHIT Consortium; Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; et al. A Human Gut Microbial Gene Catalogue Established by Metagenomic Sequencing. Nature 2010, 464, 59–65.
- Petersen, C.; Round, J.L. Defining Dysbiosis and Its Influence on Host Immunity and Disease. Cell Microbiol. 2014, 16, 1024–1033.
- Harsch, I.; Konturek, P. The Role of Gut Microbiota in Obesity and Type 2 and Type 1 Diabetes Mellitus: New Insights into “Old” Diseases. Med. Sci. 2018, 6, 32.
- Gavin, P.G.; Hamilton-Williams, E.E. The Gut Microbiota in Type 1 Diabetes: Friend or Foe? Curr. Opin. Endocrinol. Diabetes Obes. 2019, 26, 207–212.
- Zhou, H.; Sun, L.; Zhang, S.; Zhao, X.; Gang, X.; Wang, G. Evaluating the Causal Role of Gut Microbiota in Type 1 Diabetes and Its Possible Pathogenic Mechanisms. Front. Endocrinol. 2020, 11, 125.
- Atkinson, M.A. The Pathogenesis and Natural History of Type 1 Diabetes. Cold Spring Harb. Perspect. Med. 2012, 2, a007641.
- Lee, H.S.; Hwang, J.S. Genetic Aspects of Type 1 Diabetes. Ann. Pediatr. Endocrinol. Metab. 2019, 24, 143–148.
- Redondo, M.J.; Steck, A.K.; Pugliese, A. Genetics of Type 1 Diabetes. Pediatr. Diabetes 2018, 19, 346–353.
- Pociot, F. Type 1 Diabetes Genome-Wide Association Studies: Not to Be Lost in Translation. Clin. Trans. Immunol. 2017, 6, e162.
- Klak, M.; Gomółka, M.; Kowalska, P.; Cichoń, J.; Ambrożkiewicz, F.; Serwańska-Świętek, M.; Berman, A.; Wszoła, M. Type 1 Diabetes: Genes Associated with Disease Development. Cejoi 2020, 45, 439–453.
- Nyaga, D.M.; Vickers, M.H.; Jefferies, C.; Perry, J.K.; O’Sullivan, J.M. Type 1 Diabetes Mellitus-Associated Genetic Variants Contribute to Overlapping Immune Regulatory Networks. Front. Genet. 2018, 9, 535.
- Noble, J.A. Immunogenetics of Type 1 Diabetes: A Comprehensive Review. J. Autoimmun. 2015, 64, 101–112.
- Buschard, K. What Causes Type 1 Diabetes? Lessons from Animal Models. APMIS 2011, 119, 1–19.
- Oilinki, T.; Otonkoski, T.; Ilonen, J.; Knip, M.; Miettinen, P. Prevalence and Characteristics of Diabetes among Somali Children and Adolescents Living in Helsinki, Finland. Pediatr. Diabetes 2012, 13, 176–180.
- Söderström, U.; Åman, J.; Hjern, A. Being Born in Sweden Increases the Risk for Type 1 Diabetes–a Study of Migration of Children to Sweden as a Natural Experiment. Acta Paediatr. 2012, 101, 73–77.
- Rewers, M.; Ludvigsson, J. Environmental Risk Factors for Type 1 Diabetes. Lancet 2016, 387, 2340–2348.
- Esposito, S.; Toni, G.; Tascini, G.; Santi, E.; Berioli, M.G.; Principi, N. Environmental Factors Associated with Type 1 Diabetes. Front. Endocrinol. 2019, 10, 592.
- Infante, M.; Ricordi, C.; Sanchez, J.; Clare-Salzler, M.J.; Padilla, N.; Fuenmayor, V.; Chavez, C.; Alvarez, A.; Baidal, D.; Alejandro, R.; et al. Influence of Vitamin D on Islet Autoimmunity and Beta-Cell Function in Type 1 Diabetes. Nutrients 2019, 11, 2185.
- Mallone, R.; Eizirik, D.L. Presumption of Innocence for Beta Cells: Why Are They Vulnerable Autoimmune Targets in Type 1 Diabetes? Diabetologia 2020, 63, 1999–2006.
- Vaarala, O.; Atkinson, M.A.; Neu, J. The “Perfect Storm” for Type 1 Diabetes: The Complex Interplay between Intestinal Microbiota, Gut Permeability, and Mucosal Immunity. Diabetes 2008, 57, 2555–2562.
- Bibbò, S.; Dore, M.P.; Pes, G.M.; Delitala, G.; Delitala, A.P. Is There a Role for Gut Microbiota in Type 1 Diabetes Pathogenesis? Ann. Med. 2017, 49, 11–22.
- Wang, W.; Uzzau, S.; Goldblum, S.E.; Fasano, A. Human Zonulin, a Potential Modulator of Intestinal Tight Junctions. J. Cell Sci. 2000, 113 Pt 24, 4435–4440.
- Asmar, R.; Gosse, P.; Topouchian, J.; N’tela, G.; Dudley, A.; Shepherd, G.L. Effects of Telmisartan on Arterial Stiffness in Type 2 Diabetes Patients with Essential Hypertension. J. Renin. Angiotensin. Aldosterone Syst. 2002, 3, 176–180.
- Watts, T.; Berti, I.; Sapone, A.; Gerarduzzi, T.; Not, T.; Zielke, R.; Fasano, A. Role of the Intestinal Tight Junction Modulator Zonulin in the Pathogenesis of Type I Diabetes in BB Diabetic-Prone Rats. Proc. Natl. Acad. Sci. USA 2005, 102, 2916–2921.
- Fasano, A. Intestinal Permeability and Its Regulation by Zonulin: Diagnostic and Therapeutic Implications. Clin. Gastroenterol. Hepatol. 2012, 10, 1096–1100.
- Kelly, C.P.; Green, P.H.R.; Murray, J.A.; Dimarino, A.; Colatrella, A.; Leffler, D.A.; Alexander, T.; Arsenescu, R.; Leon, F.; Jiang, J.G.; et al. Larazotide Acetate in Patients with Coeliac Disease Undergoing a Gluten Challenge: A Randomised Placebo-Controlled Study. Aliment. Pharm. 2013, 37, 252–262.
- Sapone, A.; De Magistris, L.; Pietzak, M.; Clemente, M.G.; Tripathi, A.; Cucca, F.; Lampis, R.; Kryszak, D.; Cartenì, M.; Generoso, M. Zonulin Upregulation Is Associated with Increased Gut Permeability in Subjects with Type 1 Diabetes and Their Relatives. Diabetes 2006, 55, 1443–1449.
- Mønsted, M.Ø.; Falck, N.D.; Pedersen, K.; Buschard, K.; Holm, L.J.; Haupt-Jorgensen, M. Intestinal Permeability in Type 1 Diabetes: An Updated Comprehensive Overview. J. Autoimmun. 2021, 122, 102674.
- Bosi, E.; Molteni, L.; Radaelli, M.; Folini, L.; Fermo, I.; Bazzigaluppi, E.; Piemonti, L.; Pastore, M.; Paroni, R. Increased Intestinal Permeability Precedes Clinical Onset of Type 1 Diabetes. Diabetologia 2006, 49, 2824–2827.
- Secondulfo, M.; Iafusco, D.; Carratu, R.; Demagistris, L.; Sapone, A.; Generoso, M.; Mezzogiorno, A.; Sasso, F.; Cartenì, M.; De Rosa, R. Ultrastructural Mucosal Alterations and Increased Intestinal Permeability in Non-Celiac, Type I Diabetic Patients. Dig. Liver Dis. 2004, 36, 35–45.
- Paroni, R.; Fermo, I.; Molteni, L.; Folini, L.; Pastore, M.R.; Mosca, A.; Bosi, E. Lactulose and Mannitol Intestinal Permeability Detected by Capillary Electrophoresis. J. Chromatogr. B 2006, 834, 183–187.
- Maffeis, C.; Martina, A.; Corradi, M.; Quarella, S.; Nori, N.; Torriani, S.; Plebani, M.; Contreas, G.; Felis, G.E. Association between Intestinal Permeability and Faecal Microbiota Composition in Italian Children with Beta Cell Autoimmunity at Risk for Type 1 Diabetes. Diabetes/Metab. Res. Rev. 2016, 32, 700–709.
- Harbison, J.E.; Roth-Schulze, A.J.; Giles, L.C.; Tran, C.D.; Ngui, K.M.; Penno, M.A.; Thomson, R.L.; Wentworth, J.M.; Colman, P.G.; Craig, M.E.; et al. Gut Microbiome Dysbiosis and Increased Intestinal Permeability in Children with Islet Autoimmunity and Type 1 Diabetes: A Prospective Cohort Study. Pediatr. Diabetes 2019, 20, 574–583.
- Ulluwishewa, D.; Anderson, R.C.; McNabb, W.C.; Moughan, P.J.; Wells, J.M.; Roy, N.C. Regulation of Tight Junction Permeability by Intestinal Bacteria and Dietary Components. J. Nutr. 2011, 141, 769–776.
- Bedi, S.; Richardson, T.M.; Jia, B.; Saab, H.; Brinkman, F.S.L.; Westley, M. Similarities between Bacterial GAD and Human GAD65: Implications in Gut Mediated Autoimmune Type 1 Diabetes. PLoS ONE 2022, 17, e0261103.
- Jamshidi, P.; Hasanzadeh, S.; Tahvildari, A.; Farsi, Y.; Arbabi, M.; Mota, J.F.; Sechi, L.A.; Nasiri, M.J. Is There Any Association between Gut Microbiota and Type 1 Diabetes? A Systematic Review. Gut Pathog. 2019, 11, 49.
- Altindis, E.; Vomund, A.N.; Chow, I.-T.; Damasio, M.; Kwok, W.; Unanue, E.R.; Kahn, C.R. Identification of Cross Reactive Insulin Immunogenic Epitopes from Commensal Gut Microbes. Diabetes 2018, 67, 95-OR.
- Cole, D.K.; Bulek, A.M.; Dolton, G.; Schauenberg, A.J.; Szomolay, B.; Rittase, W.; Trimby, A.; Jothikumar, P.; Fuller, A.; Skowera, A.; et al. Hotspot Autoimmune T Cell Receptor Binding Underlies Pathogen and Insulin Peptide Cross-Reactivity. J. Clin. Investig. 2016, 126, 2191–2204.
- Girdhar, K.; Huang, Q.; Chow, I.-T.; Vatanen, T.; Brady, C.; Raisingani, A.; Autissier, P.; Atkinson, M.A.; Kwok, W.W.; Kahn, C.R.; et al. A Gut Microbial Peptide and Molecular Mimicry in the Pathogenesis of Type 1 Diabetes. Proc. Natl. Acad. Sci. USA 2022, 119, e2120028119.
- Hill, J.H.; Franzosa, E.A.; Huttenhower, C.; Guillemin, K. A Conserved Bacterial Protein Induces Pancreatic Beta Cell Expansion during Zebrafish Development. Elife 2016, 5, e20145.
- Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Novelo, L.L.; Casella, G.; Drew, J.C.; Ilonen, J.; Knip, M.; Hyöty, H.; et al. Toward Defining the Autoimmune Microbiome for Type 1 Diabetes. ISME J. 2011, 5, 82–91.
- de Goffau, M.C.; Luopajärvi, K.; Knip, M.; Ilonen, J.; Ruohtula, T.; Härkönen, T.; Orivuori, L.; Hakala, S.; Welling, G.W.; Harmsen, H.J.; et al. Fecal Microbiota Composition Differs between Children with β-Cell Autoimmunity and Those without. Diabetes 2013, 62, 1238–1244.
- Vatanen, T.; Franzosa, E.A.; Schwager, R.; Tripathi, S.; Arthur, T.D.; Vehik, K.; Lernmark, Å.; Hagopian, W.A.; Rewers, M.J.; She, J.-X.; et al. The Human Gut Microbiome in Early-Onset Type 1 Diabetes from the TEDDY Study. Nature 2018, 562, 589–594.
- Murri, M.; Leiva, I.; Gomez-Zumaquero, J.M.; Tinahones, F.J.; Cardona, F.; Soriguer, F.; Queipo-Ortuño, M.I. Gut Microbiota in Children with Type 1 Diabetes Differs from That in Healthy Children: A Case-Control Study. BMC Med. 2013, 11, 46.
- Alkanani, A.K.; Hara, N.; Gottlieb, P.A.; Ir, D.; Robertson, C.E.; Wagner, B.D.; Frank, D.N.; Zipris, D. Alterations in Intestinal Microbiota Correlate with Susceptibility to Type 1 Diabetes. Diabetes 2015, 64, 3510–3520.
- Kostic, A.D.; Gevers, D.; Siljander, H.; Vatanen, T.; Hyötyläinen, T.; Hämäläinen, A.-M.; Peet, A.; Tillmann, V.; Pöhö, P.; Mattila, I.; et al. The Dynamics of the Human Infant Gut Microbiome in Development and in Progression toward Type 1 Diabetes. Cell Host Microbe 2015, 17, 260–273.
- Zhang, L.; Jonscher, K.R.; Zhang, Z.; Xiong, Y.; Mueller, R.S.; Friedman, J.E.; Pan, C. Islet Autoantibody Seroconversion in Type-1 Diabetes Is Associated with Metagenome-Assembled Genomes in Infant Gut Microbiomes. Nat. Commun. 2022, 13, 3551.
- Biassoni, R.; Di Marco, E.; Squillario, M.; Barla, A.; Piccolo, G.; Ugolotti, E.; Gatti, C.; Minuto, N.; Patti, G.; Maghnie, M.; et al. Gut Microbiota in T1DM-Onset Pediatric Patients: Machine-Learning Algorithms to Classify Microorganisms as Disease Linked. J. Clin. Endocrinol. Metab. 2020, 105, dgaa407.
- Davis-Richardson, A.G.; Triplett, E.W. A Model for the Role of Gut Bacteria in the Development of Autoimmunity for Type 1 Diabetes. Diabetologia 2015, 58, 1386–1393.
- Del Chierico, F.; Conta, G.; Matteoli, M.C.; Fierabracci, A.; Reddel, S.; Macari, G.; Gardini, S.; Guarrasi, V.; Levi Mortera, S.; Marzano, V.; et al. Gut Microbiota Functional Traits, Blood PH, and Anti-GAD Antibodies Concur in the Clinical Characterization of T1D at Onset. Int. J. Mol. Sci. 2022, 23, 10256.
- van Heck, J.I.P.; Gacesa, R.; Stienstra, R.; Fu, J.; Zhernakova, A.; Harmsen, H.J.M.; Weersma, R.K.; Joosten, L.A.B.; Tack, C.J. The Gut Microbiome Composition Is Altered in Long-Standing Type 1 Diabetes and Associates with Glycemic Control and Disease-Related Complications. Diabetes Care 2022, 45, 2084–2094.
- Brown, C.T.; Davis-Richardson, A.G.; Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Casella, G.; Drew, J.C.; Ilonen, J.; Knip, M.; et al. Gut Microbiome Metagenomics Analysis Suggests a Functional Model for the Development of Autoimmunity for Type 1 Diabetes. PLoS ONE 2011, 6, e25792.
- de Goffau, M.C.; Fuentes, S.; van den Bogert, B.; Honkanen, H.; de Vos, W.M.; Welling, G.W.; Hyöty, H.; Harmsen, H.J.M. Aberrant Gut Microbiota Composition at the Onset of Type 1 Diabetes in Young Children. Diabetologia 2014, 57, 1569–1577.
- de Groot, P.F.; Belzer, C.; Aydin, Ö.; Levin, E.; Levels, J.H.; Aalvink, S.; Boot, F.; Holleman, F.; van Raalte, D.H.; Scheithauer, T.P.; et al. Distinct Fecal and Oral Microbiota Composition in Human Type 1 Diabetes, an Observational Study. PLoS ONE 2017, 12, e0188475.