Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Clinical Neurology
|
Cardiac & Cardiovascular Systems
N6-甲基腺苷(m6A)修饰是真核生物中新发现的调控机制。作为最常见的表观遗传机制之一,m6A在动脉粥样硬化(AS)和动脉粥样硬化疾病(AD)发展中的作用也越来越受到关注。
- N6-methyladenosine (m6A)
- atherosclerosis
- atherosclerotic diseases
1. Introduction
Atherosclerosis (AS) is a chronic inflammatory disease with multiple pathological features, such as endothelial dysfunction, vascular inflammation, and cholesterol accumulation. AS can cause artery plaque and stenosis, leading to the occurrence of atherosclerotic diseases (AD), such as coronary artery disease, stroke, and other arterial diseases [1,2]. AD remain the leading causes of death worldwide, and have created a vital global burden, which is still increasing [3,4]. However, the pathogenesis of AS and AD is extremely complex and largely unclear. In a word, it is of great significance to investigate the new mechanism and potential therapeutic targets of AS and AD.
A growing number of studies [5,6] show that post-transcriptional epigenetic modifications are closely related to the processes of AS and AD. N6-methyladenosine (m6A) modifications (one of the common post-transcriptional epigenetic modifications) are involved in the occurrence and development of AS and AD, and are novel and potential therapeutic targets and diagnostic biomarkers for AS and AD [7,8,9].
m6A methylation is a post-transcriptional epigenetic modification at the RNA level, which is a process of methylation of adenine at the sixth nitrogen atom catalyzed by RNA methyltransferases. m6A methylation is the most prevalent and reversible type of modification in eukaryotic mRNA, and it also plays a role in noncoding RNAs such as microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs) [10,11,12,13]. m6A methylation can regulate RNA stability, positioning, transport, splicing, and translation [14,15], which will affect the structure and function of RNAs. It plays a crucial regulatory role in the pathogenesis of various diseases, such as tumors, cardiovascular, and cerebrovascular diseases, etc. [16,17]. Recent studies [7,8,9] have identified the significant role of m6A methylation in the occurrence and development of AS and AD.
2. The Mechanisms of m6A Methylation in AS
Endothelial cells, macrophages, and smooth muscle cells are the most important initiating and developing cell types of AS. We summarized the mechanism by which m6A regulates these cell types to induce AS as follows (Figure 1).
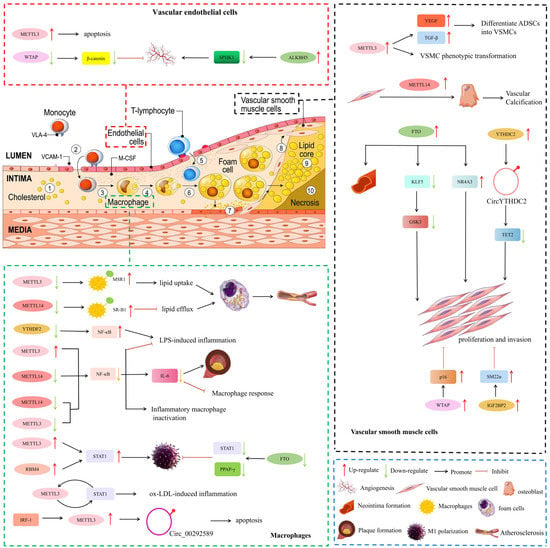
Figure 1. Potential mechanisms involved in m6A methylation-mediated regulation of AS. NR4A3, nuclear receptor subfamily 4 group A member 3; GSK3, glycogen synthase kinase 3; TET2, ten-eleven translocation 2; SM22α, smooth muscle 22α; ADSCs, adipose-derived stem cells; VSMCs, Vascular Smooth Muscle Cells; MSR1, macrophage scavenger receptor A; SR-B1, scavenger receptor B type 1; STAT1, signal transducer and activator of transcription 1; IRF-1, interferon regulatory factor-1.
2.1. Vascular Endothelial Cells
During the initial stages of AS, endothelial dysfunction and morphological damage occur, which lead to leukocyte adhesion, vasoconstriction, platelet aggregation, and thrombosis [57]. Vascular endothelial cell dysfunction is a key factor in the pathogenesis of AS.
m6A methylation plays a major role in post-transcriptional regulation in vascular endothelial cells. Zhu et al. [58] found that human cytomegalovirus (HCMV) infection could induce abnormally elevated m6A methylation, especially METTL3 and YTHDF3, leading to endothelial cell apoptosis. Wang et al. [59] used RNA transcriptome sequencing and found that in cerebral arteriovenous malformations, the expression levels of WTAP were significantly reduced but could inhibit endothelial cell angiogenesis.
m6A demethylation also plays important roles in endothelial cell angiogenesis. For example, Rajesh Kumari et al. [60] found that ALKBH5 levels were up-regulated after ischemia and correlated with the maintenance of ischemia-induced endothelial cell angiogenesis. ALKBH5 contributes to the maintenance of endothelial angiogenesis after acute ischemic stress by reducing SPHK1 m6A methylation and downstream eNOS-AKT signaling.
2.2. Macrophages Response and Inflammation
m6A methylation can influence AS progression by affecting macrophage cholesterol efflux and cell death. Cholesterol accumulation in macrophages, foam cell formation, and atherosclerotic lesions are all affected by macrophage cholesterol efflux capacity [61]. Zhao et al. [62] showed that during AS, oxidized low-density lipoprotein (ox-LDL) induced the expression of dead box protein 5 (DDX5) in macrophages and restricted the METTL3 function. METTL3 can transfer methyl groups to macrophage scavenger receptor A (MSR1) mRNA. Eventually, MSR1 mRNA stabilizes, and more MSR1 is synthesized. The uptake of more lipids further promotes the formation of foam cells, leading to the progression of AS. However, the specific mechanism of METTL3 inhibition by DDX5 is unclear. Park et al. [63] showed that MELL14 knockout attenuated cholesterol efflux and promoted foam cell formation by affecting m6A levels of scavenger receptor B type 1 (SR-B1) mRNA.
Inflammation is one of the major and fundamental pathological processes for all stages of AS [64]. The transformation of macrophages into an inflammatory phenotype is closely related to the progression of AS. Signal transducer and activator of transcription 1 (STAT1) is a key transcription factor whose activation leads to signaling cascades activated by pro-inflammatory macrophages. Liu et al. [65] showed that METTL3 has been shown to directly methylate STAT1 mRNA to increase mRNA stability, thereby upregulating STAT1 expression and promoting the polarization of M1 macrophages. Huang et al. [66] demonstrated that RBM4 regulates M1 macrophage polarization by targeting STAT1-mediated glycolysis. This study shows that RBM4 may be a candidate for regulating M1 polarization and inflammatory responses in macrophages. In addition, Gu et al. [67] found that the FTO gene knockout of m6A demethylase inhibited the phosphorylation of key proteins in the NF-κB signaling pathway, and was involved in reducing the mRNA stability of STAT1 and PPARγ through YTHDF2, thereby hindering macrophages polarization of cells. Similarly, Li et al. [68] also showed that ox-LDL stimulation significantly increased m6A-modified mRNA levels in macrophages. METTL3 promotes ox-LDL-triggered inflammation by interacting with STAT1 protein and mRNA in macrophages. In summary, m6A via the STAT1 pathway plays an important role in macrophage inflammation in AS.
In addition, m6A via the NF/κB signaling pathway also plays a crucial role in macrophage inflammation in AS. Wang et al. [69] showed that METTL3 reduced lipopolysaccharide (LPS)-induced macrophage inflammatory response by inhibiting the NF-κB pathway. However, Yu et al. [70] showed that downregulation of YTHDF2 significantly increased the LPS-induced expression of pro-inflammatory cytokines, such as IL-6 and TNF-α, and activated the MAPK and NF-κB signaling pathways. In addition, Yu et al. [71] found that the inhibition of METTL14 and METTL3 expression in macrophages could abolish m6A methylation of NF-κB mRNA, affect the stability of NF-κB mRNA, and ultimately lead to the inactivation of inflammatory macrophages, thereby significantly alleviating the progression of AS. Zheng et al. [72] found that Mettl14 knockout significantly reduced macrophage inflammatory response and atherosclerotic plaque formation through the NF-κB/IL-6 signaling pathway.
2.3. Vascular Smooth Muscle Cell (VSMC)
During AS progression, contractile VSMCs undergo phenotypic transformation into proliferative synthetic cells that generate an extracellular matrix, form fibrous caps, and stabilize plaques [75]. Accumulating evidence suggests that m6A can affect the pathophysiological function of VSMCs.
The “writers” of m6A methylation are involved in VSMC proliferation and migration. Lin et al. [76] showed that hypoxia can affect METTL3 expression and further affect m6A modification of related factors such as VEGF and TGF-β, thereby inducing adipose-derived stem cells (ADSCs) to differentiate into VSMCs. Chen et al. [77] found that overexpressed METTL14 increased m6A methylation by promoting the transformation of VSMCs to osteoblasts, and played an important role in the pathological mechanism of vascular calcification. Furthermore, in the study by Zhu et al. [78], the expression of WTAP in VSMCs altered cell proliferation and migration. Total notoginseng saponins regulate p16 m6A methylation by promoting WTAP expression, thereby inhibiting intimal thickening.
The “erasers” of m6A demethylases have been reported to promote VSMC proliferation and migration. For example, Ma et al. [79] demonstrated that both FTO overexpression and Ang II-induced FTO expression promoted VSMC proliferation and migration. FTO promotes the expression of Kruppel-like factor 5 (KLF5) mRNA by reducing the m6A methylation of KLF5 mRNA, thereby upregulating the expression of downstream glycogen synthase kinase 3 (GSK3). Similarly, Huo et al. [80] also showed that FTO promoted Ang II-induced VSMC proliferation and inflammatory response by demethylating the m6A methylation of nuclear receptor subfamily 4 group A member 3 (NR4A3) mRNA.
3. The Role of m6A Methylation in AS and AD
m6A methylation not only causes the most common atherosclerotic diseases (such as coronary heart disease (CHD) and ischemic stroke (IS) through AS. Moreover, m6A also plays an important role in the injury and repair of CHD and IS. In Table 1, the progress of m6A methylation regulator-guided epigenetic modification in AS and AD.
Table 1. m6A methylation regulator-guided epigenetic modification in AS and AD.
| Atherosclerotic Process | m6A Regulators | Expression | Target Gene | Main Function | Reference |
|---|---|---|---|---|---|
| AS | METTL3 | ↑ | LRP6 and DVL1 | Enhances translation of LRP6 and DVL1, modulates Wnt signaling, and thus exerts angiogenic effects | [84] |
| METTL3 | ↑ | PGC-1α mRNA | Promotes mitochondrial dysfunction and ox-LDL-induced inflammation | [73] | |
| METTL3 | ↓ | JAK2/STAT3 | Alleviates ox-LDL-induced endothelial cell dysfunction, prevents in vivo angiogenesis of developing embryos, and hinders progression in AS mice models | [85] | |
| METTL3 | ↑ | NLRP1 and KLF4 | Up-regulates NLRP1, down-regulates KLF4, hypermethylates m6A, and triggers atherosclerotic response | [86] | |
| METTL3 | ↑ | miR-375-3p/PDK1 | Makes AS plaques more vulnerable | [87] | |
| METTL3 | ↑ | EGFR | Promotes EGFR degradation and alleviates endothelial atherogenic progression | [88] | |
| METTL14 | ↑ | FOXO1 | Increases FOXO1 m6A methylation, aggravates endothelial inflammation and AS | [89] | |
| METTL14 | ↓ | miR-19a | Inhibits the proliferation and invasion of ASVEC | [90] | |
| METTL14 | ↑ | LncRNA ZFAS1 | Plays a vital role in AS | [91] | |
| METTL14 | ↓ | p65 mRNA | Relieves the development of AS | [92] | |
| METTL14 | ↓ | NF-κB/IL-6 | Reduces the inflammation response of macrophages and the development of AS plaques | [72] | |
| FTO | ↑ | Not known | Modulates neointima formation in vivo | [81] | |
| FTO | ↓ | NR4A3 | Alleviates AngII-induced VSMC proliferation and inflammatory response | [80] | |
| ALKBH5 | ↓ | HIF1α | Inhibits the expression of MIAT induced by ox-LDL | [42] | |
| CHD | METTL3 | ↑ | TFEB | Promotes cardiomyocyte apoptosis | [8] |
| WTAP | ↑ | ATF4 | Promotes endoplasmic reticulum stress and apoptosis, aggravates myocardial I/R injury | [93] | |
| FTO | ↑ | SERCA2A MYH6/7 RYR2 | Reverses ischemic damage | [94] | |
| FTO | ↑ | MHRT | Inhibits cardiomyocyte apoptosis | [95] | |
| ALKBH5 | ↓ | TFEB | Promotes cardiomyocyte apoptosis | [8] | |
| ALKBH5 | ↑ | WNT5A | Regulates angiogenesis after ischemia | [96] | |
| IS | METTL3 | ↑ | miR-335 | Promotes formation of SG and reduces damage of IS | [97] |
| YTHDC1 | ↑ | Not known | Protects rats from brain damage | [98] |
3.1. AS
AS is the main cause of CHD and IS [99]. Quiles Jiménez et al. [6] used mass spectrometry to analyze m6A methylation levels in tissue from non-atherosclerotic arterial and carotid atherosclerotic patients, which showed the changes in the expression levels of m6A writers, erasers, and readers in atherosclerotic tissue. The findings of Wu et al. [42] showed that m6A methylation levels were significantly reduced in peripheral blood leukocytes of atherosclerotic patients and mice. The bioinformatic analysis indicated that differentially methylated genes were involved in the pathogenesis of AS. These findings suggest that m6A methylation is involved in the occurrence and progression of AS.
METTL3-dependent m6A methylation was recently shown to play an important role in AS. For example, Yao et al. [84] demonstrated that METTL3 promotes the translation of low-density lipoprotein receptor-related protein 6 (LRP6) and dishevelled 1 (DVL1) in human umbilical vein endothelial cells (HUVEC) under hypoxic stress in a YTHDF1-dependent manner, thereby exerting an angiogenesis effect. Zhang et al. [73] demonstrated that METTL3 plays a role in ox-LDL-induced monocyte inflammation, in which METTL3 and YTHDF2 synergistically modify PGC-1α mRNA, mediate its degradation, and reduce PGC-1α protein levels, thereby enhancing the inflammatory response. This study provides new insights into the role of METTL3-dependent m6A methylation of PGC-1α mRNA in the inflammatory response of monocytes. In addition, Dong et al. [85] explored the role and molecular mechanism of m6A-METTL3 in AS progression from an in vivo perspective using an AS mouse model and an in vivo chick embryo chorioallantoic membrane assay. The results indicated that METTL3 knockout prevented AS progression through IGF2BP1 inhibition of the JAK2/STAT3 pathway. Furthermore, Chien et al. [86] showed that METTL3 up-regulated NOD-like receptor protein 1 (NLRP1) and down-regulated Kruppel-like factor 4 (KLF4) in an m6A-dependent manner. METTL3 exerts pro-inflammatory effects in HUVEC or mouse aortic endothelial cells exposed to pro-atherosclerotic oscillatory stress or TNF-α stimulation, thereby promoting inflammatory cell adhesion and AS pathogenesis. In a recent study, Chen et al. [87] found that silencing METTL3 alleviated AS progression in mice. Silencing METTL3 suppressed m6A levels and decreased the binding of DGCR8 to pri-miR-375, further limiting the expression of miR-375-3p. miR-375-3p targets PDK1 transcription. Ultimately silencing METTL3 plays a role in stabilizing AS plaques. However, Li et al. [88] reported a protective role of METTL3 in AS. The authors found that METTL3 promotes m6A-dependent degradation of epidermal growth factor receptor (EGFR) mRNA, a molecule associated with vascular endothelial cell (EC) dysfunction, thereby attenuating the progression of endothelial atherosclerosis.
Similarly, METTL14 also plays a pivotal role in the process of AS. Jian et al. [89] constructed a model of EC inflammation induced by TNF-α. With an increase in the expression of METTL14 in endothelial cells stimulated with TNF-α, METTL14 increases the m6A methylation of FOXO1, promoting its expression, which triggers endothelial inflammatory responses and the development of AS. Subsequent in vivo experiments showed that METTL14 knockout could inhibit AS plaque development in an m6A-dependent manner in METTL14 knockout mice. Furthermore, Zhang et al. [90] pointed out that METTL14 promoted the production of mature miR-19a by increasing the expression of m6A in miR-19a, thereby accelerating the invasion and proliferation of cardiovascular ECs. Chen et al. [100] showed that the m6A methylation of Zinc finger NFX type 1 (ZNFX1) antisense RNA 1 (ZFAS1) was significantly higher in AS patients than in controls, and that m6A methylation in ZFAS1 was regulated by METTl14. Tang et al. [91] found that METTl14 affects the expression of downstream ADAM10/RAB22A by affecting the m6A methylation of LncRNA ZFAS1, thereby participating in cholesterol metabolism and vascular inflammation, and ultimately regulating the occurrence and development of AS. Liu et al. [92] demonstrated that silencing METTL14 attenuates the development of AS through the m6A methylation of p65 mRNA by establishing an in vitro atherosclerotic cell model and an in vivo high-fat diet mouse model. Zheng et al. [72] showed that Mettl14 plays a crucial role in macrophage inflammation in AS through the NF-κB/IL-6 signaling pathway. METTL14 knockout significantly reduced the macrophage inflammatory response and atherosclerotic plaque formation.
3.2. AD
3.2.1. CHD
CHD, also known as ischemic cardiomyopathy, refers to the clinical syndrome of long-term myocardial ischemia caused by coronary atherosclerosis, resulting in diffuse myocardial fibrosis [101].
Mathiyalagan et al. [94] demonstrated for the first time that mRNA m6A methylation was significantly higher in ischemic myocardium than in non-ischemic regions. Song et al. [8] demonstrated that m6A RNA methylation is involved in the development of myocardial hypoxia/reperfusion injury by regulating autophagy. Deng et al. [81] identified differentially methylated m6A sites in mRNAs and lncRNAs between peripheral blood mononuclear cells of the CHD group and control group. These studies suggest that m6A RNA methylation plays a crucial role in CHD.
Song et al. [8] established a mice model of hypoxia-reperfusion and ischemia-reperfusion and found that the m6A methylation levels in mice cardiomyocytes increased, and METTL3 is the main cause of abnormal modification of m6甲基化。沉默METTL3可增强自噬通量并抑制缺氧/复氧心肌细胞中的心肌细胞凋亡。然而,METTL3的过表达或抑制m6脱甲基酶ALKBH5促进心肌细胞凋亡。这表明METTL3是自噬的负调节因子。同样,WTAP 通过增加 mRNA m 来促进内质网 (ER) 应激和凋亡6ATF4(一种控制ER相关基因表达并上调其表达的转录因子)水平,从而加重心肌I/R损伤[93]。
FTO介导的m6去甲基化也与心肌 I/R 损伤有关。FTO可以选择性去甲基化肌质网Ca。2+-ATP酶(SERCA2A),肌球蛋白重链6/7(MYH6/7),ryanodine受体2(RYR2)和其他影响心脏钙稳态,肌原纤维合成和收缩功能的mRNA。它通过 m 增加上述基因的转录和翻译6A依赖性通路,从而逆转缺血性损伤[94]。肌球蛋白重链相关RNA转录本(MHRT)是一种源自MYH7基因反义链的心脏特异性lncRNA。Shen等人[95]证明,FTO的过表达通过降低6MHRT的修改。
此外,ALKBH5的过表达可以逆转METTL3对心肌细胞的破坏作用[8]。另一项研究探讨了ALKBH5对缺血后血管生成的影响和机制。Zhao等人[96]证明ALKBH5通过降低WNT家族成员5A(WNT5A)mRNA的mA水平并促进其在心脏微血管内皮细胞中的降解来负调节缺血后的血管生成。
3.2.2. 是
Si等人[97]通过使用大鼠大脑中动脉闭塞模型在原代皮质神经元和PC12细胞中建立了氧-葡萄糖剥夺/再灌注模型,以探索大鼠的氧-葡萄糖剥夺/再灌注模型。6急性缺血性卒中早期阶段参与应力颗粒 (SG) 形成的潜在机制的甲基化。体外和体内结果均显示,METTL3蛋白、m6随着再灌注时间的延长,A水平和miR-335表达显著降低。该发现表明,METTL3介导的m6甲基化在促进SG形成和减少疾病早期阶段的IS损伤中起重要作用。
与METTL3类似,YTHDC1在IS的病理过程中也起着保护作用。Zhang等[98]发现YTHDC1的敲除加重了缺血性脑损伤,而YTHDC1的过表达保护了大鼠免受脑损伤;在机制上,YTHDC1促进PTEN mRNA降解以增加Akt磷酸化,从而促进神经元存活,特别是在缺血后。
This entry is adapted from the peer-reviewed paper 10.3390/jcdd9110367
This entry is offline, you can click here to edit this entry!