Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Obstetrics & Gynaecology
Placental mesenchymal dysplasia (PMD) is a morphological abnormality resembling partial hydatidiform moles without abnormal trophoblastic proliferation. In PMD, approximately 20% of fetuses have Beckwith–Wiedemann syndrome (BWS), and approximately 20% of BWS fetuses are associated with PMD. In addition, PMD is a cardinal feature of BWS, and paternal uniparental diploidy/biparental diploidy mosaicism (also called androgenetic/biparental mosaicism) has been found in both BWS and PMD.
- placental mesenchymal dysplasia
- genomic imprinting
- androgenetic/biparental mosaicism (paternal uniparental diploidy/biparental diploidy mosaicism)
1. Introduction
Placental mesenchymal dysplasia (PMD) was first reported by Moscoso [1] as diffuse mesenchymal hyperplasia of the placental stem villi, leading to increased placental volume (placentomegaly) and elevated levels of alpha fetoprotein (AFP). It is now known to be characterized by varying levels of placentomegaly, aneurysmally dilated chorionic plate vessels, thrombosis of the dilated vessels, and large grapelike vesicles within the placenta [2]. Since these features may mimic a molar pregnancy on ultrasound, it is often mistaken for partial hydatidiform mole (PHM) or complete hydatidiform mole (CHM) with a coexisting normal fetus [3]. Unlike molar pregnancies, PMD usually features a normal fetus and the pregnancy often extends into the third trimester [4]. However, PMD pregnancies are high-risk because they carry fixed probabilities of several complications. These include fetal growth restriction (FGR); preterm delivery; fetal demise; preeclampsia; eclampsia; hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome; and hypertensive disorders of pregnancy (HDP) [2,3,5,6]. In addition, approximately 20% of fetuses in PMD pregnancies have Beckwith–Wiedemann syndrome (BWS) [4]. Since androgenetic/biparental mosaicism (ABM) and androgenetic/biparental chimera (ABC) have been found in PMD specimens, the presence of androgenetic cells resulting in abnormal genomic imprinting has been suggested as a cause of PMD [7,8,9,10].
BWS (OMIM #130650) was originally reported by Beckwith and Wiedemann [11,12] as a syndrome involving exomphalos, macroglossia, and gigantism. It is now known to be an imprinting disorder with complex and diverse phenotypes. Macroglossia, exomphalos, and lateralized overgrowth are the cardinal features, and there is also an increased risk of developing embryonal tumors such as Wilms tumor, hepatoblastoma, neuroblastoma, rhabdomyosarcoma, adrenocortical carcinoma, or phaeochromocytoma [13,14]. Because of the range of phenotypes, it is recommended that the Beckwith–Wiedemann spectrum (BWSp) is used and that diagnoses are made based on a scoring system. This system assigns two points to each cardinal feature and one point to each suggestive feature. Patients with a total score of ≥4 are diagnosed as classical BWS, irrespective of their molecular test results, as are patients with a score of ≥2 with positive genetic test results [13]. Placentomegaly and PMD are included as the suggestive or cardinal feature, respectively, in the BWSp scoring system [13]. There are five major causative alterations: loss of methylation of imprinting control region 2 (KCNQ1OT1:TSS-differentially methylated region (DMR)) (ICR2-LOM), gain of methylation at ICR1 (H19/IGF2:IG-DMR) (ICR1-GOM), paternal uniparental disomy of 11 (pUPD11), loss-of-function variants of the CDKN1C gene, and paternal duplication of 11p15 [13]. The genetic testing aims to detect these major alterations. Several minor alterations have also been discovered thus far: genetic variants within ICR1, paternal uniparental diploidy/biparental diploidy mosaicism (PUDM, also called ABM), and genetic variants of the KCNQ1 gene [15,16,17,18,19].
2. Incidence and Pathology of PMD
2.1. Incidence
In 2001, Paradinas et al. reported the incidence of PMD as 0.2% (15 out of 7560 placentas examined) [21]. The following year, Arizawa et al. reported an incidence of only 0.02% (7/30,758) [22], and in 2012, Zeng et al. reported an incidence of 0.002% (2/95,265) [23]. Although the intermediate figure (0.02%) is widely used in the literature, calculation of the true incidence may be difficult, since only a small fraction of placentas are subjected to pathologic examination [3]. In any case, PMD is certainly a rare placental condition, and so far only just over 100 cases have been reported [3,24].
More than 80% of the fetuses from PMD pregnancies identified thus far have been female with the normal karyotype (46,XX) [3,4,6]. However, a karyotype of 46,XX/46,XY has been observed in some fetuses. Cohen et al. reported a normal karyotype in 32 of 36 cases (89%) and an abnormal karyotype in the remaining four (11%) [20]. The abnormal karyotypes in that study included trisomy 13, 47,XXY (Klinefelter syndrome), and 69,XXX (triploidy), and these karyotypes were confirmed in the placental specimens. In addition, 13q12.11 deletion in a neonate and another case of trisomy 13 in a fetus have been reported, but these were not confirmed in PMD specimens [25,26].
2.2. Pathology
Macroscopically, PMD is usually characterized by placentomegaly, meaning that the weight of the placenta is greater than the 95th percentile of placenta weights [27], and is often associated with dilatation and congestion of the vessels in the chorionic plate. In some cases, grape-like cysts formed by dilatation of the hydropic stem villi are present (Figure 1A) [5]. Most placentas with PMD have two distinct areas: one with a macroscopically normal appearance and one exhibiting macroscopic PMD characteristics (a cystic lesion) (Figure 1A) [3,28]. In a small proportion of cases the macroscopic PMD lesion occupies the entire placenta.
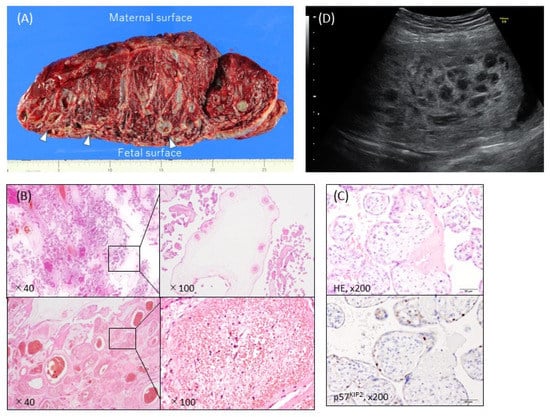
Figure 1. Typical ultrasonographic and macroscopic views and histopathological findings of placental mesenchymal dysplasia (PMD). (A) Macroscopic view of the incised plane. The placental weight was 1530 g at 38 weeks and 0 days of pregnancy. There were heterogeneous areas, with numerous cysts containing gelatinous liquid (arrows) and normal red-brown or spongy villous tissue. The cystic areas were predominantly on the fetal placental surface. (B) Hematoxylin-eosin stain. The PMD featured enlarged edematous stem villi with cisterns. Dilated, thick-walled, and thrombosed blood vessels with fibromuscular hyperplasia were also apparent. No trophoblastic proliferation was observed. (C) Immunohistochemistry against p57KIP2, which was not expressed in the stroma cells of dysplastic villi in the PMD placenta. (D) Ultrasonography revealed a thickened chorionic plate with a multicystic lesion and the living fetus (19 weeks and 2 days of pregnancy).
The microscopic features of PMD include enlarged stem-cell villi with varying degrees of edema and containing abnormal thick-walled vessels, which in some cases are thrombosed (Figure 1B). Trophoblastic proliferation and stromal inclusions, both characteristic of molar pregnancies, are absent (Figure 1B) [5]. The product of the maternally expressed imprinted gene CDKN1C, p57KIP2, is strongly expressed in the cytotrophoblasts and villous mesenchyme in normal placentas, but in CHM it is absent or markedly reduced, regardless of whether it is androgenetic or biparental CHM (BiCHM) [29,30,31]. In PMD, p57KIP2 expression is absent in the stromal cells (Figure 1C), which contain the androgenetic genome, but normal in the villous cytotrophoblast cells, which contain the biparental genome, leading to normal proliferation [8,32,33].
3. Clinical Findings in PMD
3.1. Ultrasound Findings
In ultrasound imaging, PMD is usually detected as a cystic placenta that has hypoechoic areas (80%), is enlarged or thickened (50%), and has dilated chorionic villi (16%) (Figure 1D) [5]. The majority of 49 PMD cases showed a thickened chorionic plate with a multicystic lesion that resembled PHM or CHM with co-twin during the first half of the pregnancy [6]. The course of the cystic lesions was varied: in some cases they gradually became more apparent and in others they disappeared [6].
3.2. Maternal Complications and Pregnancy and Neonatal Outcome
Maternal serum α-fetoprotein (MSAFP) was elevated in 70–80% of patients with PMD, while maternal serum human chorionic gonadotropin (MShCG) levels for the gestational age were elevated in 18–38% of patients with PMD [5,6]. For prenatal diagnosis, in addition to ultrasound findings, elevated MSAFP levels with normal MShCG levels in the second and third trimesters may be indicative of PMD [6].
Nine percent of PMD cases in one study showed preeclampsia, eclampsia, HELLP, or gestational hypertension as maternal complications [5]. In another study, hypertensive disorders of pregnancy (HDP) were found in 12.8% of PMD cases [6].
Fetal growth restriction (FGR), preterm delivery, and fetal demise occurred in respectively 33–72%, 53–64%, and 13–18% of cases covered in recent PMD case reviews [2,5,6]. Neonatal outcomes include BWS, hepatic tumors, and hematologic disorders (anemia and thrombocytopenia) [3,5,6]. In the following we discuss BWS and hepatic tumors.
4. Complications of PMD
4.1. BWS
BWS is frequently associated with PMD. Infants delivered from mothers with PMD were diagnosed as BWS in 12 of 66 (18.2%), 15 of 71 (21.1%), 12 of 64 (18.8%), and 8 of 47 PMD cases (17.0%), respectively, in four previous reports [4,5,6,20]. Conversely, PMD was a complication in the pregnancies of 13 of 60 neonates with BWS (21.7%) [34]. In our own data, we recorded seven PMD pregnancies among 66 neonates with BWS (10.6%). Therefore, the frequencies of BWS in PMD cases and of PMD in BWS cases are roughly similar.
Placentomegaly was observed in 70.9% and 12.0% of BWS patients in two reports, which suggests using this as a suggestive feature in the BWSp scoring system [34,35]. In the placental pathological findings in one of these studies, there was no significant difference between the causative molecular subgroups for BWS [34]. Armes et al. performed pathological examinations on eight placentas: three enlarged ones from BWS patients with ICR2-LOM, one PMD one from a BWS patient with pUPD11, and four PMD placentas without BWS features [32]. They did not find any pathological features of PMD in the three placentas from the BWS patients with ICR2-LOM, but they did find a striking excess of extravillous trophoblast. Since four of the PMD placentas exhibited ABM, and a fifth had pUPD11, they concluded that IGF2 was a strong candidate gene for PMD.
4.2. Hepatic Mesenchymal Hamartoma
Hepatic mesenchymal hamartoma (HMH) is defined as the excessive, focal growth of an admixture of hepatic vascular and epithelial components, which becomes multicystic as it enlarges [36]. It is the second-most-common benign liver tumor in children [37] and one of the most commonly occurring tumors in fetuses from PMD pregnancies. An HMH case that was probably complicated with PMD, since it showed prominent dilatation of the placental vessels with multiple thromboses, was reported in 1983 [38]. Thus far, 19 HMH cases with PMD have been reported [23,36,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55]. Of these, three cases showed ABM in both the HMH and the PMD [45,51,52], one showed ABM in the HMH but had no genotype information for the PMD [53], and one showed ABM in the PMD but had no genotype information for the HMH [55]. Based on these findings and the recurrent identification of ABM in both sporadic PMD and sporadic HMH, ABM resulting in the imbalanced expression of imprinted genes has been considered as a causative mechanism [45,52]. For example, expression of C19MC, which is a placenta-specific imprinted locus that encodes a cluster of 46 microRNAs (miRNAs), increased in 10 of 10 cases of sporadic HMH and three cases of HMH associated with ABM, despite the absence of expression in the normal liver [52]. In another study, cases with ABM-associated HMH and some cases of sporadic HMH harbored a hypomethylated allele of the C19MC differentially methylated region 1 (C19MC-DMR1, also called DPRX/MIR512:IG-DMR) located at its promoter region [53,56]. In addition, chromosomal rearrangement or translocation involving 19q13.4, the locus of C19MC, may disrupt the C19MC promoter and drive the expression of C19MC miRNAs [53]. These observations suggest the involvement of increased expression of the C19MC miRNA cluster as a result of hypomethylation of the C19MC-DMR1 (DPRX/MIR512:IG-DMR) or rearrangement of the C19MC promoter in the pathogenesis of HMH. However, since other cases with HMH did not express C19MC miRNAs, there may also be causative molecular mechanisms that are independent of C19MC [53].
This entry is adapted from the peer-reviewed paper 10.3390/cancers14225563
This entry is offline, you can click here to edit this entry!