Next-generation sequencing (NGS) is widely used to study microorganisms, allowing the elucidation of bacteria and viruses inhabiting different body systems and identifying new pathogens.
- metagenomics
- virome
- DNA/RNA
- virus
1. Introduction
Along with classical diagnostic methods such as virus isolation, serology, and PCR, NGS plays an important role in virus identification, especially in outbreaks of known and/or new diseases [1].
NGS technology is currently revolutionizing the field of genomics and clinical virology is no exception. High-throughput sequencing techniques have made significant contributions to many areas of virology, including virus discovery and metagenomics, molecular epidemiology, pathogenesis, and research into how viruses evade the host’s immune response. Previously, unknown viruses have been identified using NGS techniques, including a new rhabdovirus associated with acute hemorrhagic fever identified in Central Africa [2] and a new cyclovirus found in the cerebrospinal fluid of patients with infections of the central nervous system [3].
Metagenomics refers to the study of the complete genomic composition of a complex mixture of microorganisms [69]. Unlike bacteria, viruses do not have a common gene for all families, and therefore the study of the virome is based on complex analytical methods. In addition to detecting viruses, NGS is also capable of providing additional information on virulence markers, epidemiology, genotyping, and evolution of pathogens, as well as estimating the copy number from the number of DNA/RNA reads [4][5].
NGS methods are also used to study the genetic and phenotypic heterogeneity of viruses during replication in host cells, which is missed in conventional studies using consensus sequencing data. An example of such a study is Ikuyo Takayama (2021), who studied the genetic diversity of the A(H1N1)pdm09 virus using NGS in upper and lower respiratory tract samples from nine patients. Significant genetic heterogeneity was found in the samples, including 47 amino acid substitutions and 1 D222G/N substitution in the hemagglutinin was common to several patients. The authors note the need for such studies in order to not miss the potentially important mutations that occur during viral replication in the host, especially in patients with severe disease [6]. The NGS process typically consists of two parts. The first was experimental work in the laboratory (wet lab), including the stages of sample preparation, DNA/RNA extraction, library preparation, and sequencing. The second part is bioinformatic analysis (dry lab), which includes data quality control, removal of non-target DNA, and analysis of nucleotide sequences [7].
The application of NGS to viral studies has certain experimental and analytical features, in contrast to the study of microbial communities.
This applies to sample preparation and sequencing. For example, it must be considered that viral genomes (especially RNA) are rather fragile and easily destroyed and the ratio of viral material to the host genome is very low (less than 1%). Therefore, an important procedure is to conduct the procedure of amplification of target viral nucleic acids or enrichment of viral particles [8][4].
In some cases, it is necessary to reverse-transcribe the viral RNA before PCR and sequencing. PCR amplification leads to errors that are difficult to distinguish from real mutations. In addition to PCR errors, all NGS platforms introduce sequencing errors at a rate similar to the mutation rate in RNA viruses [9]. Additionally, the depth of sequencing, that is the number of unique reads including a given nucleotide in the sequence, can also vary depending on the genome [8].
Modern viral NGS protocols have already been optimized for detecting both RNA and DNA viruses [10][11][12]. In addition, viral particle enrichment techniques are often used to increase the relative concentration of viral particles and/or their nucleotide sequences, as well as methods of depleting host genomic DNA and ribosomal or mRNA. These methods are laborious and not easy to automate for routine use in clinical diagnostics, which imposes restrictions on their use in mass clinical diagnostics [4].
Difficulties in the detection of viruses by NGS in clinical specimens, especially respiratory ones, are due to the presence of an extremely small number of viruses and their nucleic acids in the study samples, compared with the high content of host genomic material and the bacterial component. These circumstances determine the high importance of the preliminary steps in NGS sequencing. Proper sampling and production of nucleic sequences of interest are critical for obtaining the desired results (Figure).
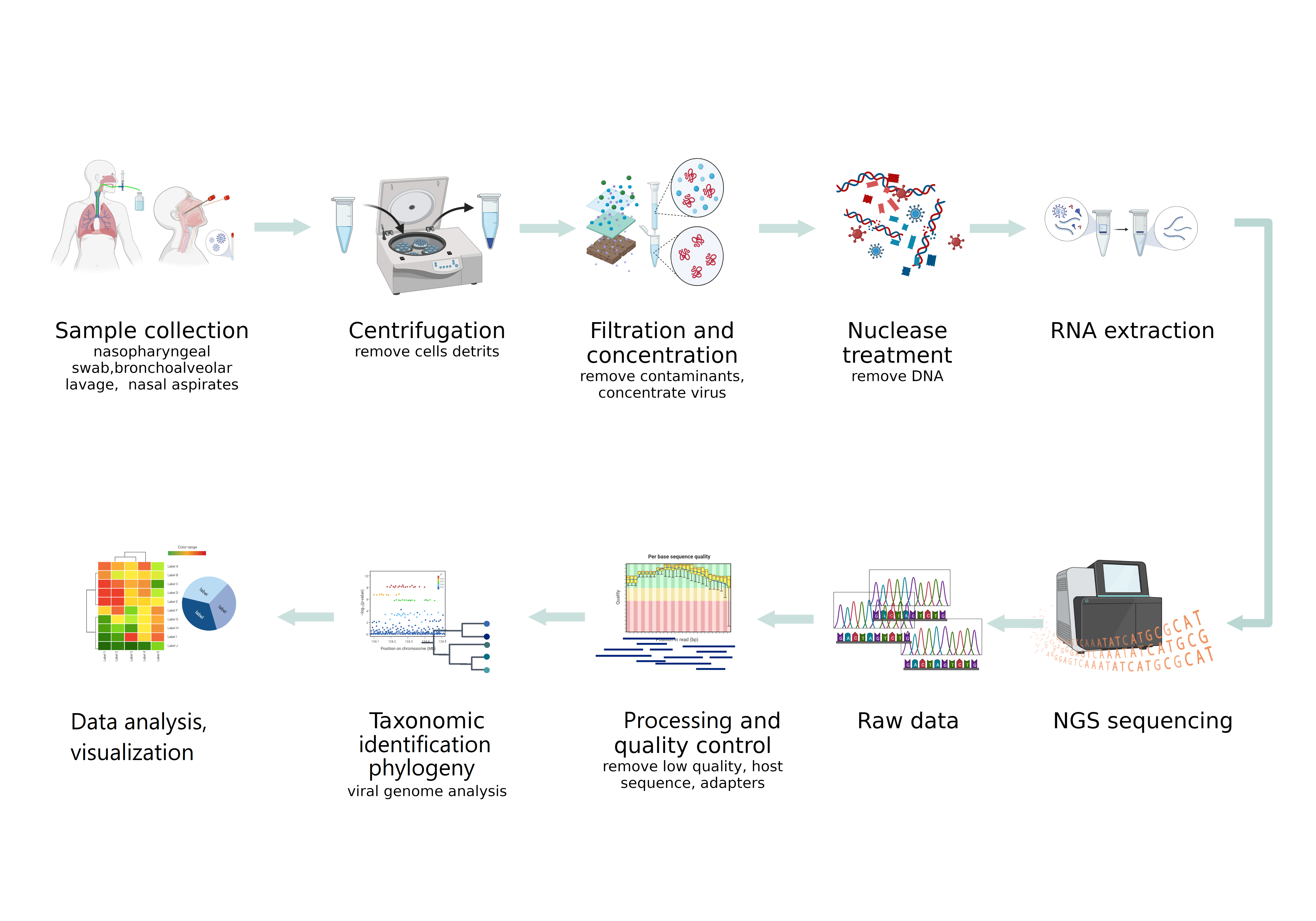
3. NGS Platforms for Virome Studies
Initially, metagenomics was actively developed in the study of bacterial genomes and achieved tremendous success. However, a new direction in metagenomics, the virome has been actively developed. Virus studies using NGS methods are now at the peak of their development and technological approaches are being improved every time. Examples of applications are pathogen detection, including novel detection, species identification, and typing, detection of antibiotic resistance, virulence, and more [1][13][14].
With the development of NGS, its practical application is constantly expanding, especially in clinical virology in the diagnosis of new or previously undetected pathogens of infectious diseases [8][15][16]. It was shown that the sensitivity of the NGS method is comparable to that of the PCR method with increasing sequencing depth [17][1].
So, W.I. Lipkin (2013) in his works because of the bioinformatic analysis, revealed new viruses, such as rhabdovirus associated with acute hemorrhagic fever and cyclovirus found in the cerebrospinal fluid of patients [18][19]. Using the NGS method T. Kustin detected human parainfluenza 1 virus, human parainfluenza 4 virus, and influenza C among 54 patients [20].
In 91 samples of NFS by the NGS method were identified human rhinoviruses, enteroviruses, influenza A virus, coronavirus OC43 and respiratory syncytial virus (RSV) A, as well as rotavirus, torque teno virus, human papillomavirus, human betaherpesvirus 7, cyclovirus, vientovirus, gemycircularvirus and statovirus [21]. When examining NFS in 48 children, NGS revealed 11 RNA viruses, 4 DNA viruses, 4 bacterial species, and one fungus [22].
H. Mostafa (2020) in studies, when detecting SARS-CoV-2 by NGS in 500 patients, showed the possibility of diagnosing other infections and analyzing the respiratory microbiome [15].
Yi-Yi Qian (2021) showed that the sensitivity of NGS turned out to be higher than that of the traditional cultivation method, but in comparison with PCR, these indicators were lower [16]. Thorburn (2015) studied 89 nasopharyngeal swabs the sensitivity and specificity of the NGS method compared to Real-Time PCR were 78% and 80%, respectively [23]. So, the NGS technology as a diagnostic tool is still in the development stage, and approaches to its application are being improved every year.
Technically, NGS is run on various platforms, which can be divided by reading length into short-read and long-read. The short-read sequencing approaches fall into two categories: sequencing by ligation (SBL) and sequencing by synthesis (SBS).
In most approaches, SBL and SBS DNA are clonally amplified on a solid surface. The presence of many thousands of identical copies of a piece of DNA in a certain area ensures that the signal can be distinguished from background noise. Mass parallelization is also facilitated by the creation of many millions of individual SBL or SBS reaction centers, each with its own clonal DNA template. The sequencing platform can simultaneously collect information from many millions of reaction sites, thereby sequencing many millions of DNA molecules in parallel.
SBL technologies include Applied Biosystems/SOLiD and MGI/BGI/Complete Genomics. Sequencing by synthesis (SBS) is performed on the Illumina and Qiagen platforms.
Illumina offers a popular series of sequencing platforms–ISeq, MiSeq, MiniSeq, NextSeq, HiSeq, and NovaSeq. High throughput and low error rate (less than 1%) are the main reasons why this technology currently dominates the field of virology and beyond.
The very first NGS platform for studying viral metagenomics was Life Science/Roche 454, a pyrosequencing method. The 454 sequencing has been widely used to identify several new viruses and virome profiles from human and animal samples [24], including arboviruses [25], orbiviruses [26], arenaviruses [27], Lujo virus [28], astrovirus [29], gyroviruses [30], porcine bocaviruses [31], picornaviruses [32], rhabdoviruses [2], coronaviruses [33], gamma papillomavirus [34], and seadornavirus [35]. Most of these viruses have been identified in serum, respiratory, and feces samples.
Although this technology offered a higher yield than Sanger sequencing at a lower cost, this technology has been supplanted by other NGS technologies due to its high cost, errors in homopolymer regions, and low throughput.
Ion Torrent semiconductor sequencing technology with the Ion Proton and Ion S5 series sequencers which benefits from fast sequencing makes these sequencers particularly useful for targeted detection of viruses in clinical specimens, such as HIV [36], hepatitis B virus [37], HCV [38] and rapid genome sequencing of several viruses, including Tuscany virus [39], polyomavirus [40], porcine reproductive and respiratory syndrome virus [41], orthoreovirus [42], bluetongue virus [43], rotavirus [44], influenza virus [45]. This technology has been used to study the virome of skin [46], ticks [47], intestines in piglets [48] and seals [49].
Over the past few years, sequencing technologies have grown rapidly, introducing of third-generation sequencing (TGS) technologies such as Oxford Nanopore and PacBio platforms which are real-time single molecule sequencing (SMRT), that which reduces amplification bias and short reading problems. The reduction in cost and time presented by these sequencing methods is a valuable benefit.
TGS is considered the next revolution in sequencing technology. Sequencing of long sequences and speed, without PCR amplification, allows uniform coverage of the entire genome. This technology has also been used for virus sequencing [50][51][52]. Looking forward, future developments in TGS should focus on improving sequencing accuracy and high throughput.
4. Bioinformatics and NGS Data Analysis
An important stage in metagenomics is computer analysis or bioinformatics, the task is to process a big array of NGS data, which can be represented by sequences of the genomes of viruses, bacteria, humans, animals, and others.
When using early sequencing methods, sequences are usually classified using NCBI BLAST [53] against the NCBI (nt) database[54]. However, when using NGS data, it is necessary to process a much larger number of short (up to 300 bps) reads, for which homologous regions are not always available in databases and possible sequencing errors made by the sequencer must be considered.
Therefore, NGS needs specialized methods of analysis. Many biological information specialists have developed computational workflows for the analysis of viral metagenomes. Their publications describe many computer tools for taxonomic classification. While these tools can be helpful, choosing the right workflow can be difficult, especially for less experienced users [55][56].
Bioinformatics involves the processing of sequencing data for checking the quality of reads, filtering sequences, and their identification. Some of the workflows of metagenomics have been tested and described in review articles [57][58][59][60][61].
There are specialized programs and online services for virus analysis, such as Viral MetaGenome Annotation Pipeline (VMGAP) [62][63], Viral Informatics Resource for Metagenomic Exploration (VIROME) [64] and Metavir 2 [65], DisCVR [66].
Additionally, there are cloud-deployed clinical metagenomic computing workflows such as SURPI (sequence-based ultra-rapid pathogen identification) [67] and CZ ID (IDseq) [68], for the detection and identification of pathogens.
The CosmosID program has been used to analyze the microbiome of various groups and quantify microorganisms [69][70][71].
Annotated data visualization programs are available for MEGAN, Pavian, Krona, PanViz, MetaViz, and Anvi’o. MEGAN and Pavian perform broad analyses but require specific inputs that make them less suitable for different workflows. PanViz, MetaViz, and Anvi’o are sharpened for the analysis of bacteria and are of little use for viruses. The available programs Geneious and CLC bio are paid for and require an expensive license [72][73][74][75][76].
To separate viral and non-viral sequences, vFams is used [77]. The VIP program is also used to identify viruses [35]. Virus-TAP, VirusSeker for BLAST-based virus identification with modules (VS-VIROME and VS-DISCOVERY), and SHIVER for de novo assembly [78][79][80].
In the age of NGS and bioinformatics, open, easily accessible, free, and globally distributed platforms for data analysis can significantly change the accessibility and quality of biomedical research. Baker et al. (2020) showed the possibility and importance of data exchange using the example of SARS-CoV-2 [81]. For example, for all virus genomic data, the Galaxy platform (https://usegalaxy.eu/ accessed on 18 November 2022) was used, which can be replicated using open-source tools by any researcher with an Internet connection.
The opportunities for such access allow for raising community awareness in the absence of primary data needed to respond to global emergencies such as the COVID-19 outbreak effectively, transparently, and reproducibly perform all analyzes on an equal footing.
Additionally, the publication emphasizes the problem of non-reproducibility of results that are published in scientific papers and, which cannot be completed again because the data are not shared or deliberately hidden. Thus, any researcher should be able to apply the same analytical procedures to their data and have access to all data analysis tools, including computing power and infrastructure [81].
This entry is adapted from the peer-reviewed paper 10.3390/microorganisms10122327
References
- Quiñones-Mateu, M.E.; Avila, S.; Reyes-Teran, G.; Martinez, M.A.; Deep Sequencing: Becoming a Critical Tool in Clinical Virology. J. Clin. Virol. 2014, 61, 9–19, https://doi.org/10.1016/j.jcv.2014.06.013.
- Grard, G.; Fair, J.N.; Lee, D.; Slikas, E.; Steffen, I.; Muyembe, J.-J.; Sittler, T.; Veeraraghavan, N.; Ruby, J.G.; Wang, C.; et al. A Novel Rhabdovirus Associated with Acute Hemorrhagic Fever in Central Africa. PLoS Pathog. 2012, 8, e1002924, https://doi.org/10.1371/journal.ppat.1002924.
- 68. Van Tan, L.; van Doorn, H.R.; Nghia, H.D.T.; Chau, T.T.H.; Tu, L.T.P.; de Vries, M.; Canuti, M.; Deijs, M.; Jebbink, M.F.; Baker, S.; et al. Identification of a New Cyclovirus in Cerebrospinal Fluid of Patients with Acute Central Nervous System Infections. mBio 2013, 4, e00231-13, doi.org/10.1128/mBio.00231-13.
- van Boheemen, S.; van Rijn, A.L.; Pappas, N.; Carbo, E.C.; Vorderman, R.H.P.; Sidorov, I.; van’t Hof, P.J.; Mei, H.; Claas, E.C.J.; Kroes, A.C.M.; et al. Retrospective Validation of a Metagenomic Sequencing Protocol for Combined Detection of RNA and DNA Viruses Using Respiratory Samples from Pediatric Patients. J. Mol. Diagn 2020, 22, 196–207, https://doi.org/10.1016/j.jmoldx.2019.10.007.
- Mongkolrattanothai, K.; Naccache, S.N.; Bender, J.M.; Samayoa, E.; Pham, E.; Yu, G.; Dien Bard, J.; Miller, S.; Aldrovandi, G.; Chiu, C.Y.; et al. Neurobrucellosis: Unexpected Answer From Metagenomic Next-Generation Sequencing. J. Pediatr. Infect. Dis. Soc. 2017, 6, 393–398, https://doi.org/10.1093/jpids/piw066.
- Takayama, I.; Nguyen, B.G.; Dao, C.X.; Pham, T.T.; Dang, T.Q.; Truong, P.T.; Van Do, T.; Pham, T.T.P.; Fujisaki, S.; Odagiri, T.; et al. Next-Generation Sequencing Analysis of the Within-Host Genetic Diversity of Influenza A(H1N1)Pdm09 Viruses in the Upper and Lower Respiratory Tracts of Patients with Severe Influenza.. mSphere 2021, 6, e01043-20, https://doi.org/10.1128/mSphere.01043-20.
- Li, N.; Cai, Q.; Miao, Q.; Song, Z.; Fang, Y.; Hu, B.; High‐Throughput Metagenomics for Identification of Pathogens in the Clinical Settings. Small Methods 2021, 5, 2000792, https://doi.org/10.1002/smtd.202000792.
- Pérez-Losada, M.; Arenas, M.; Galán, J.C.; Bracho, M.A.; Hillung, J.; García-González, N.; González-Candelas, F.; High-Throughput Sequencing (HTS) for the Analysis of Viral Populations. Infect. Genet. Evol. 2020, 80, 104208, https://doi.org/10.1016/j.meegid.2020.104208.
- Sanjuán, R.. Viral Mutation Rates. In Virus Evolution: Current Research and Future Directions; Caister Academic Press: Norfolk, UK, 2016; pp. 1–28.
- Kohl, C.; Brinkmann, A.; Dabrowski, P.W.; Radonić, A.; Nitsche, A.; Kurth, A.; Protocol for Metagenomic Virus Detection in Clinical Specimens. Emerg. Infect. Dis. 2015, 21, 48–57, https://doi.org/10.3201/eid2101.140766.
- Parker, J.; Chen, J.; Application of next Generation Sequencing for the Detection of Human Viral Pathogens in Clinical Specimens. J. Clin. Virol. 2017, 86, 20-26, https://doi.org/10.1016/j.jcv.2016.11.010.
- Zou, X.; Tang, G.; Zhao, X.; Huang, Y.; Chen, T.; Lei, M.; Chen, W.; Yang, L.; Zhu, W.; Zhuang, L.; et al. Simultaneous Virus Identification and Characterization of Severe Unexplained Pneumonia Cases Using a Metagenomics Sequencing Technique. Sci. China Life Sci. 2017, 60, 279–286, https://doi.org/10.1007/s11427-016-0244-8.
- Bharucha, T.; Oeser, C.; Balloux, F.; Brown, J.R.; Carbo, E.C.; Charlett, A.; Chiu, C.Y.; Claas, E.C.J.; de Goffau, M.C.; de Vries, J.J.C.; et al. STROBE-Metagenomics: A STROBE Extension Statement to Guide the Reporting of Metagenomics Studies. Lancet Infect. Dis. 2020, 20, e251–e260, https://doi.org/10.1016/S1473-3099(20)30199-7.
- Capobianchi, M.R.; Giombini, E.; Rozera, G.; Next-Generation Sequencing Technology in Clinical Virology. Clin. Microbiol. Infect. 2013, 19, 15–22, https://doi.org/10.1111/1469-0691.12056.
- Mostafa, H.H.; Fissel, J.A.; Fanelli, B.; Bergman, Y.; Gniazdowski, V.; Dadlani, M.; Carroll, K.C.; Colwell, R.R.; Simner, P.J.; Metagenomic Next-Generation Sequencing of Nasopharyngeal Specimens Collected from Confirmed and Suspect COVID-19 Patients. mBio 2020, 11, e01969-20, https://doi.org/10.1128/mBio.01969-20.
- Qian, Y.-Y.; Wang, H.-Y.; Zhou, Y.; Zhang, H.-C.; Zhu, Y.-M.; Zhou, X.; Ying, Y.; Cui, P.; Wu, H.-L.; Zhang, W.-H.; et al. Improving Pulmonary Infection Diagnosis with Metagenomic Next Generation Sequencing. Front. Cell. Infect. Microbiol. 2021, 10, 567615, https://doi.org/10.3389/fcimb.2020.567615.
- Lecuit, M.; Eloit, M.; The Human Virome: New Tools and Concepts. Trends Microbiol. 2013, 21, 510–515, https://doi.org/10.1016/j.tim.2013.07.001.
- Lipkin, W.I.; Firth, C.; Viral Surveillance and Discovery. Curr. Opin. Virol. 2013, 3, 199–204, https://doi.org/10.1016/j.coviro.2013.03.010.
- del Campo, J.A.; Parra-Sánchez, M.; Figueruela, B.; García-Rey, S.; Quer, J.; Gregori, J.; Bernal, S.; Grande, L.; Palomares, J.C.; Romero-Gómez, M.; et al. Hepatitis C Virus Deep Sequencing for Sub-Genotype Identification in Mixed Infections: A Real-Life Experience. Int. J. Infect. Dis. 2018, 67, 114–117, https://doi.org/10.1016/j.ijid.2017.12.016.
- Kustin, T.; Ling, G.; Sharabi, S.; Ram, D.; Friedman, N.; Zuckerman, N.; Bucris, E.D.; Glatman-Freedman, A.; Stern, A.; Man-delboim, M.; et al. A Method to Identify Respiratory Virus Infections in Clinical Samples Using Next-Generation Sequencing. Sci. Rep. 2019, 9, 2206, https://doi.org/10.1038/s41598-018-37483-w.
- Thi Kha Tu, N.; Thi Thu Hong, N.; Thi Han Ny, N.; My Phuc, T.; Thi Thanh Tam, P.; van Doorn, H.R.; Dang Trung Nghia, H.; Thao Huong, D.; An Han, D.; Thi Thu Ha, L.; et al. The Virome of Acute Respiratory Diseases in Individuals at Risk of Zoonotic Infections. Viruses 2020, 12, 960, https://doi.org/10.3390/v12090960.
- Li, C.-X.; Li, W.; Zhou, J.; Zhang, B.; Feng, Y.; Xu, C.-P.; Lu, Y.-Y.; Holmes, E.C.; Shi, M.; High Resolution Metagenomic Characterization of Complex Infectomes in Paediatric Acute Respiratory Infection. Sci. Rep. 2020, 10, 3963, https://doi.org/10.1038/s41598-020-60992-6.
- Thorburn, F.; Bennett, S.; Modha, S.; Murdoch, D.; Gunson, R.; Murcia, P.R.; The Use of next Generation Sequencing in the Diagnosis and Typing of Respiratory Infections. J. Clin. Virol. 2015, 69, 96–100, https://doi.org/10.1016/j.jcv.2015.06.082.
- Day, J.M.; Ballard, L.L.; Duke, M.V.; Scheffler, B.E.; Zsak, L.; Metagenomic Analysis of the Turkey Gut RNA Virus Community. Virol. J. 2010, 7, 313, https://doi.org/10.1186/1743-422X-7-313.
- Bishop-Lilly, K.A.; Turell, M.J.; Willner, K.M.; Butani, A.; Nolan, N.M.E.; Lentz, S.M.; Akmal, A.; Mateczun, A.; Brahmbhatt, T.N.; Sozhamannan, S.; et al. Arbovirus Detection in Insect Vectors by Rapid, High-Throughput Pyrosequencing. PLoS Negl. Trop. Dis. 2010, 4, e878, https://doi.org/10.1371/journal.pntd.0000878.
- Li, Y.; Fu, X.; Ma, J.; Zhang, J.; Hu, Y.; Dong, W.; Wan, Z.; Li, Q.; Kuang, Y.-Q.; Lan, K.; et al. Altered Respiratory Virome and Serum Cytokine Profile Associated with Recurrent Respiratory Tract Infections in Chil-dren. Nat. Commun. 2019, 10, 2288, https://doi.org/10.1038/s41467-019-10294-x.
- Palacios, G.; Druce, J.; Du, L.; Tran, T.; Birch, C.; Briese, T.; Conlan, S.; Quan, P.-L.; Hui, J.; Marshall, J.; et al. A New Arenavirus in a Cluster of Fatal Transplant-Associated Diseases. N. Engl. J. Med. 2008, 358, 991–998, https://doi.org/10.1056/NEJMoa073785.
- Briese, T.; Paweska, J.T.; McMullan, L.K.; Hutchison, S.K.; Street, C.; Palacios, G.; Khristova, M.L.; Weyer, J.; Swanepoel, R.; Egholm, M.; et al. Genetic Detection and Characterization of Lujo Virus, a New Hemorrhagic Fever–Associated Arenavirus from Southern Africa. PLoS Pathog. 2009, 5, e1000455, https://doi.org/10.1371/journal.ppat.1000455.
- Quan, P.-L.; Wagner, T.A.; Briese, T.; Torgerson, T.R.; Hornig, M..; Tashmukhamedova, A.; Firth, C.; Palacios, G.; Baisre-De-Leon, A.; Paddock, C.D.; et al. Astrovirus Encephalitis in Boy with X-Linked Agammaglobulinemia. Emerg. Infect. Dis. 2010, 16, 918–925, https://doi.org/10.3201/eid1606.091536.
- Phan, T.G.; Li, L.; O’Ryan, M.G.; Cortes, H.; Mamani, N.; Bonkoungou, I.J.O.; Wang, C.; Leutenegger, C.M.; Delwart, E.; A Third Gyrovirus Species in Human Faeces. J. Gen. Virol. 2012, 93, 1356–1361, https://doi.org/10.1099/vir.0.041731-0.
- Yu, J.; Li, J.; Ao, Y.; Duan, Z.; Detection of Novel Viruses in Porcine Fecal Samples from China. Virol. J. 2013, 10, 39, https://doi.org/10.1186/1743-422X-10-39.
- Boros, Á.; Nemes, C.; Pankovics, P.; Kapusinszky, B.; Delwart, E.; Reuter, G.; Identification and Complete Genome Characterization of a Novel Picornavirus in Turkey (Meleagris gallopavo). J. Gen. Virol. 2012, 93, 2171–2182, https://doi.org/10.1099/vir.0.043224-0.
- Honkavuori, K.S.; Briese, T.; Krauss, S.; Sanchez, M.D.; Jain, K.; Hutchison, S.K.; Webster, R.G.; Lipkin, W.I.; Novel Coronavirus and Astrovirus in Delaware Bay Shorebirds. PLoS ONE 2014, 9, e93395, https://doi.org/10.1371/journal.pone.0093395.
- Phan, T.G.; Vo, N.P.; Aronen, M.; Jartti, L.; Jartti, T.; Delwart, E.; Novel Human Gammapapillomavirus Species in a Nasal Swab. Genome Announc. 2013, 1, e00022-13, https://doi.org/10.1128/genomeA.00022-13.
- Reuter, G.; Boros, Á.; Delwart, E.; Pankovics, P.; Novel Seadornavirus (Family Reoviridae) Related to Banna Virus in Europe. Arch. Virol 2013, 158, 2163–2167, https://doi.org/10.1007/s00705-013-1712-9.
- Gibson, R.M.; Meyer, A.M.; Winner, D.; Archer, J.; Feyertag, F.; Ruiz-Mateos, E.; Leal, M.; Robertson, D.L.; Schmotzer, C.L.; Quiñones-Mateu, M.E.; et al. Sensitive Deep-Sequencing-Based HIV-1 Genotyping Assay to Simultaneously Determine Susceptibility to Protease, Re-verse Transcriptase, Integrase, and Maturation Inhibitors, as Well as HIV-1 Coreceptor Tropism. Antimicrob. Agents Chemother. 2014, 58, 2167–2185, https://doi.org/10.1128/AAC.02710-13.
- Yan, L.; Zhang, H.; Ma, H.; Liu, D.; Li, W.; Kang, Y.; Yang, R.; Wang, J.; He, G.; Xie, X.; et al. Deep Sequencing of Hepatitis B Virus Basal Core Promoter and Precore Mutants in HBeAg-Positive Chronic Hepatitis B Patients. Sci. Rep. 2015, 5, 17950, https://doi.org/10.1038/srep17950.
- Gaspareto, K.V.; Ribeiro, R.M.; de Mello Malta, F.; Gomes-Gouvêa, M.S.; Muto, N.H.; Romano, C.M.; Mendes-Correa, M.C.; Carrilho, F.J.; Sabino, E.C.; Pinho, J.R.R.; et al. Resistance-Associated Variants in HCV Subtypes 1a and 1b Detected by Ion Torrent Sequencing Platform. Antivir. Ther. 2016, 21, 653–660, https://doi.org/10.3851/IMP3057.
- Nougairede, A.; Bichaud, L.; Thiberville, S.-D.; Ninove, L.; Zandotti, C.; de Lamballerie, X.; Brouqui, P.; Charrel, R.N.; Isolation of Toscana Virus from the Cerebrospinal Fluid of a Man with Meningitis in Marseille, France, 2010. Vector-Borne Zoonotic Dis. 2013, 13, 685–688, https://doi.org/10.1089/vbz.2013.1316.
- Anthony, S.J.; St. Leger, J.A.; Navarrete-Macias, I.; Nilson, E.; Sanchez-Leon, M.; Liang, E.; Seimon, T.; Jain, K.; Karesh, W.; Daszak, P.; et al. Identification of a Novel Cetacean Polyomavirus from a Common Dolphin (Delphinus delphis) with Tracheobronchitis. PLoS ONE 2013, 8, e68239, https://doi.org/10.1371/journal.pone.0068239.
- Kvisgaard, L.K.; Hjulsager, C.K.; Fahnøe, U.; Breum, S.Ø.; Ait-Ali, T.; Larsen, L.E.; A Fast and Robust Method for Full Genome Sequencing of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) Type 1 and Type 2. J. Virol. Methods 2013, 193, 697–705, https://doi.org/10.1016/j.jviromet.2013.07.019.
- Steyer, A.; Gutiérrez-Aguire, I.; Kolenc, M.; Koren, S.; Kutnjak, D.; Pokorn, M.; Poljšak-Prijatelj, M.; Rački, N.; Ravnikar, M.; Sagadin, M.; et al. High Similarity of Novel Orthoreovirus Detected in a Child Hospitalized with Acute Gastroenteritis to Mammalian Or-thoreoviruses Found in Bats in Europe. J. Clin. Microbiol. 2013, 51, 3818–3825, https://doi.org/10.1128/JCM.01531-13.
- Lorusso, A.; Marcacci, M.; Ancora, M.; Mangone, I.; Leone, A.; Marini, V.; Cammà, C.; Savini, G.; Complete Genome Sequence of Bluetongue Virus Serotype 1 Circulating in Italy, Obtained through a Fast Next-Generation Sequencing Protocol. Genome Announc. 2014, 2, e00093-14, https://doi.org/10.1128/genomeA.00093-14.
- Ndze, V.N.; Esona, M.D.; Achidi, E.A.; Gonsu, K.H.; Dóró, R.; Marton, S.; Farkas, S.; Ngeng, M.B.; Ngu, A.F.; Obama-Abena, M.T.; et al. Full Genome Characterization of Human Rotavirus A Strains Isolated in Cameroon, 2010–2011: Diverse Combinations of the G and P Genes and Lack of Reassortment of the Backbone Genes. Infect. Genet. Evol. 2014, 28, 537–560, doi.org/10.1016/j.meegid.2014.10.009.
- Van den Hoecke, S.; Verhelst, J.; Vuylsteke, M.; Saelens, X.; Analysis of the Genetic Diversity of Influenza A Viruses Using Next-Generation DNA Sequencing. BMC Genom. 2015, 16, 79, https://doi.org/10.1186/s12864-015-1284-z.
- Bzhalava, D.; Johansson, H.; Ekström, J.; Faust, H.; Möller, B.; Eklund, C.; Nordin, P.; Stenquist, B.; Paoli, J.; Persson, B.; et al. Unbiased Approach for Virus Detection in Skin Lesions. PLoS ONE 2013, 8, e65953, https://doi.org/10.1371/journal.pone.0065953.
- Xia, H.; Hu, C.; Zhang, D.; Tang, S.; Zhang, Z.; Kou, Z.; Fan, Z.; Bente, D.; Zeng, C.; Li, T.; et al. Metagenomic Profile of the Viral Communities in Rhipicephalus Spp. Ticks from Yunnan, China. PLoS ONE 2015, 10, e0121609, https://doi.org/10.1371/journal.pone.0121609.
- Karlsson, O.E.; Larsson, J.; Hayer, J.; Berg, M.; Jacobson, M. T.; The Intestinal Eukaryotic Virome in Healthy and Diarrhoeic Neonatal Piglets. PLoS ONE 2016, 11, e0151481, https://doi.org/10.1371/journal.pone.0151481.
- Kluge, M.; Campos, F.S.; Tavares, M.; de Amorim, D.B.; Valdez, F.P.; Giongo, A.; Roehe, P.M.; Franco, A.C.; Metagenomic Survey of Viral Diversity Obtained from Feces of Subantarctic and South American Fur Seals. PLoS ONE 2016, 11, e0151921, https://doi.org/10.1371/journal.pone.0151921.
- Warwick-Dugdale, J.; Solonenko, N.; Moore, K.; Chittick, L.; Gregory, A.C.; Allen, M.J.; Sullivan, M.B.; Temperton, B.; Long-Read Viral Metagenomics Captures Abundant and Microdiverse Viral Populations and Their Niche-Defining Ge-nomic Islands. PeerJ 2019, 7, e6800, https://doi.org/10.7717/peerj.6800.
- Naveca, F.G.; Claro, I.; Giovanetti, M.; de Jesus, J.G.; Xavier, J.; Iani, F.C.d.M.; do Nascimento, V.A.; de Souza, V.C.; Silveira, P.P.; Lourenço, J.; et al. Genomic, Epidemiological and Digital Surveillance of Chikungunya Virus in the Brazilian Amazon. PLoS Negl. Trop. Dis. 2019, 13, e0007065, https://doi.org/10.1371/journal.pntd.0007065.
- Mohsin, H.; Asif, A.; Fatima, M.; Rehman, Y.; Potential Role of Viral Metagenomics as a Surveillance Tool for the Early Detection of Emerging Novel Pathogens. Arch. Microbiol. 2021, 203, 865–872, https://doi.org/10.1007/s00203-020-02105-5.
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J.; Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410, https://doi.org/10.1016/S0022-2836(05)80360-2.
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2022, 50, D20–D26, https://doi.org/10.1093/nar/gkab1112.
- Posada-Cespedes, S.; Seifert, D.; Beerenwinkel, N.; Recent Advances in Inferring Viral Diversity from High-Throughput Sequencing Data. Virus Res. 2017, 239, 17-32, https://doi.org/10.1016/j.virusres.2016.09.016.
- Rose, R.; Constantinides, B.; Tapinos, A.; Robertson, D.L.; Prosperi, M.; Challenges in the Analysis of Viral Metagenomes. Virus Evol. 2016, 2, vew022, https://doi.org/10.1093/ve/vew022.
- Taş, N.; de Jong, A.E.; Li, Y.; Trubl, G.; Xue, Y.; Dove, N.C.; Metagenomic Tools in Microbial Ecology Research. Curr. Opin. Biotechnol. 2021, 67, 184–191, https://doi.org/10.1016/j.copbio.2021.01.019.
- Nooij, S.; Schmitz, D.; Vennema, H.; Kroneman, A.; Koopmans, M.P.G.; Overview of Virus Metagenomic Classification Methods and Their Biological Applications. Front. Microbiol. 2018, 9, 749, https://doi.org/10.3389/fmicb.2018.00749.
- Yang, C.; Chowdhury, D.; Zhang, Z.; Cheung, W.K.; Lu, A.; Bian, Z.; Zhang, L. A .; Review of Computational Tools for Gener-ating Metagenome-Assembled Genomes from Metagenomic Sequencing Data. Comput. Struct. Biotechnol. 2021, 19, 6301–6314, https://doi.org/10.1016/j.csbj.2021.11.028.
- Kayani, M.u.R.; Huang, W.; Feng, R.; Chen, L.; Genome-Resolved Metagenomics Using Environmental and Clinical Samples. Brief. Bioinform. 2021, 12, bbab030, https://doi.org/10.1093/bib/bbab030.
- Sharma, D.; Priyadarshini, P.; Vrati, S.; Unraveling the Web of Viroinformatics: Computational Tools and Databases in Virus Research. . J. Virol. 2015, 89, 1489–1501, https://doi.org/10.1128/JVI.02027-14.
- Metsky, H.C.; Siddle, K.J.; Gladden-Young, A.; Qu, J.; Yang, D.K.; Brehio, P.; Goldfarb, A.; Piantadosi, A.; Wohl, S.; Carter, A.; et al. Capturing Sequence Diversity in Metagenomes with Comprehensive and Scalable Probe Design. Nat. Biotechnol. 2019, 37, 160–168 , https://doi.org/10.1038/s41587-018-0006-x.
- Lorenzi, H. . Viral Metagenome Annotation Pipeline. In Encyclopedia of Metagenomics; Springer: New York: New York, NY, USA, 2013; pp. 1–12.
- Wommack, K.E.; Bhavsar, J.; Polson, S.W.; Chen, J.; Dumas, M.; Srinivasiah, S.; Furman, M.; Jamindar, S.; Nasko, D.J.; VIROME: A Standard Operating Procedure for Analysis of Viral Metagenome Sequences. Stand. Genom. Sci. 2012, 6, 427–439 , https://doi.org/10.4056/sigs.2945050.
- Roux, S.; Tournayre, J.; Mahul, A.; Debroas, D.; Enault, F.; Metavir 2: New Tools for Viral Metagenome Comparison and As-sembled Virome Analysis. BMC Bioinform. 2014, 15, 76, https://doi.org/10.1186/1471-2105-15-76.
- Maabar, M.; Davison, A.J.; Vučak, M.; Thorburn, F.; Murcia, P.R.; Gunson, R.; Palmarini, M.; Hughes, J.; DisCVR: Rapid Viral Diagnosis from High-Throughput Sequencing Data. Virus Evol. 2019, 5, vez033, https://doi.org/10.1093/ve/vez033.
- Naccache, S.N.; Federman, S.; Veeraraghavan, N.; Zaharia, M.; Lee, D.; Samayoa, E.; Bouquet, J.; Greninger, A.L.; Luk, K.-C.; Enge, B.; et al. A Cloud-Compatible Bioinformatics Pipeline for Ultrarapid Pathogen Identification from next-Generation Sequencing of Clinical Samples. Genome Res. 2014, 24, 1180–1192, https://doi.org/10.1101/gr.171934.113.
- Kalantar, K.L.; Carvalho, T.; de Bourcy, C.F.A.; Dimitrov, B.; Dingle, G.; Egger, R.; Han, J.; Holmes, O.B.; Juan, Y.-F.; King, R.; et al. IDseq—An Open Source Cloud-Based Pipeline and Analysis Service for Metagenomic Pathogen Detection and Monitoring. Gigascience 2020, 9, giaa111, https://doi.org/10.1093/gigascience/giaa111.
- Fitzpatrick, A.H.; Rupnik, A.; O’Shea, H.; Crispie, F.; Keaveney, S.; Cotter, P.; High Throughput Sequencing for the Detection and Characterization of RNA Viruses. Front. Microbiol. 2021, 12, 621719, https://doi.org/10.3389/fmicb.2021.621719.
- Hasan, N.A.; Young, B.A.; Minard-Smith, A.T.; Saeed, K.; Li, H.; Heizer, E.M.; McMillan, N.J.; Isom, R.; Abdullah, A.S.; Bornman, D.M.; et al. Microbial Community Profiling of Human Saliva Using Shotgun Metagenomic Sequencing. PLoS ONE 2014, 9, e97699 , https://doi.org/10.1371/journal.pone.0097699.
- Ponnusamy, D.; Kozlova, E.V.; Sha, J.; Erova, T.E.; Azar, S.R.; Fitts, E.C.; Kirtley, M.L.; Tiner, B.L.; Andersson, J.A.; Grim, C.J.; et al. Cross-Talk among Flesh-Eating Aeromonas Hydrophila Strains in Mixed Infection Leading to Necrotizing Fasciitis. Proc. Natl. Acad. Sci. USA 2016, 113, 722–727, https://doi.org/10.1073/pnas.1523817113.
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C.; MEGAN Analysis of Metagenomic Data. Genome Res. 2007, 17, 377–386, https://doi.org/10.1101/gr.5969107.
- Breitwieser, F.P.; Salzberg, S.L.; Pavian: Interactive Analysis of Metagenomics Data for Microbiome Studies and Pathogen Identification. Bioinformatics 2020, 36, 1303–1304, https://doi.org/10.1093/bioinformatics/btz715.
- Pedersen, T.L.; Nookaew, I.; Wayne Ussery, D.; Månsson, M.; PanViz: Interactive Visualization of the Structure of Function-ally Annotated Pangenomes. Bioinformatics 2017, 33, 1081–1082, https://doi.org/10.1093/bioinformatics/btw761.
- Wagner, J.; Chelaru, F.; Kancherla, J.; Paulson, J.N.; Zhang, A.; Felix, V.; Mahurkar, A.; Elmqvist, N.; Corrada Bravo, H.; Metaviz: Interactive Statistical and Visual Analysis of Metagenomic Data. Nucleic Acids Res. 2018, 46, 2777–2787 , https://doi.org/10.1093/nar/gky136.
- Eren, A.M.; Esen, Ö.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O.; Anvi’o: An Advanced Analysis and Visualization Platform for ‘omics Data. PeerJ 2015, 3, e1319, https://doi.org/10.7717/peerj.1319.
- Skewes-Cox, P.; Sharpton, T.J.; Pollard, K.S.; DeRisi, J.L.; Profile Hidden Markov Models for the Detection of Viruses within Metagenomic Sequence Data. PLoS ONE 2014, 9, e105067, https://doi.org/10.1371/journal.pone.0105067.
- Yamashita, A.; Sekizuka, T.; Kuroda, M.; VirusTAP: Viral Genome-Targeted Assembly Pipeline. Front. Microbiol. 2016, 7, 32, https://doi.org/10.3389/fmicb.2016.00032.
- Lin, J.; Kramna, L.; Autio, R.; Hyöty, H.; Nykter, M.; Cinek, O.; Vipie: Web Pipeline for Parallel Characterization of Viral Populations from Multiple NGS Samples. BMC Genom. 2017, 18, 378, https://doi.org/10.1186/s12864-017-3721-7.
- Lin, H.-H.; Liao, Y.-C..; DrVM: A New Tool for Efficient Genome Assembly of Known Eukaryotic Viruses from Meta-genomes. Gigascience 2017, 6, gix003, https://doi.org/10.1093/gigascience/gix003.
- Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Chilton, J.; Coraor, N.; Coppens, F.; Eguinoa, I.; Gladman, S.; Grüning, B.; et al. No More Business as Usual: Agile and Effective Responses to Emerging Pathogen Threats Require Open Data and Open Analytics. PLoS Pathog. 2020, 16, e1008643, https://doi.org/10.1371/journal.ppat.1008643.