Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Placental growth factor (PlGF) and its receptor, fms-like tyrosine kinase-1 (Flt-1), are important regulators involved in angiogenesis, atherogenesis, and inflammation. Elevation of circulating soluble isoform of Flt-1 (sFlt-1) and downregulation of sFlt-1 in the vascular endothelium by uremic toxins and oxidative stress both exacerbate heart failure and atherosclerosis. Circulating sFlt-1 is inconsistent with sFlt-1 synthesis, because levels of matrix-bound sFlt-1 are much higher than those of circulating sFlt-1, as verified by a heparin loading test, and are drastically reduced in chronic kidney disease (CKD).
- PlGF
- sFlt-1
- cardiorenal connection
1. Introduction
The number of patients with chronic kidney disease (CKD) has been increasing globally, and these individuals currently comprise more than 10% of the general population in developed countries [1]. CKD is associated with two critical risks: the progression of end-stage renal disease and the development of cardiovascular diseases. The death rate due to cardiovascular diseases exceeds the rate of progression to more severe nephropathy in diabetic kidney disease [2]. The coexistence of cardiovascular and renal disease is associated with a higher risk of cardiovascular events and death than the presence of either condition alone [3]. Organ cross-talk between the heart and kidneys is widely recognized, and is referred to as the “cardiorenal connection” [4]. The molecular mechanisms underlying this phenomenon are not fully understood, but the natriuretic peptide family and fibroblast growth factors have been established as leading players in the pathogenesis of cardiac hypertrophy and vascular calcification in CKD [5][6]. Recently, however, the cardiorenal connection has been shown to be increasingly complex since several underlying mechanisms have been identified; these include endothelial injury, immunological imbalance, cell death, inflammatory cascades, and oxidative stress [7].
2. PlGF/Flt-1 Pathway
2.1. PlGF
The human vascular endothelial growth factor (VEGF) family consists of two groups of molecules: VEGF-A, VEGF-B, and placental growth factor (PlGF), which are primarily involved in the growth of blood vessels [8]; and VEGF-C and VEGF-D, which contribute mainly to the growth of lymphatic vessels [9]. VEGF ligands can form anti-parallel homodimers and heterodimers that optimize binding to these ligands’ preferred receptors and facilitate receptor dimerization [10].
VEGF-associated receptors (VEGFR-1, also known as Flt-1; VEGFR-2, also known as Flk-1 in mice and KDR in humans; and VEGFR-3, also known as Flt-4) are characterized by a ligand-binding region with seven immunoglobulin-like domains, a single transmembrane domain, and a tyrosine kinase domain with a long kinase insert (Figure 1). VEGF-A, VEGF-C, and VEGF-D bind to Flt-1 and Flk-1, while PlGF and VEGF-B bind only to Flt-1. The VEGF family plays a wide variety of crucial roles in the regulation of angiogenesis, lymphangiogenesis, lipid metabolism, inflammation, atherosclerosis, and tumor development [11][12].

Figure 1. VEGF receptors and a soluble isoform of VEGFR-1. The VEGF isoforms have three receptors (VEGFR-1, VEGFR-2, and VEGFR-3) that contain a tyrosine–kinase domain and an extracellular region with seven Ig-like loops. The soluble isoform of Flt-1 consists of extracellular domains with motifs that bind to VEGF-A, PlGF, and heparin.
Two years after VEGF was identified, PlGF was first discovered in the human placenta, where it is highly expressed in trophoblasts and endothelial cells and promotes placental growth [13]. PlGF was later found to be expressed in many other organs, such as the heart, lungs, and brain [14][15][16]. Unlike VEGF, PlGF is redundant for vascular development and physiological vessel maintenance in healthy adults, and it is highly expressed in pathological conditions such as malignancy, tissue ischemia, and inflammation [17]. Experimental studies showed that local delivery of the PlGF gene into the carotid artery of hypercholesterolemic rabbits enhanced intimal thickening via macrophage accumulation [18]. Moreover, treatment with a neutralizing monoclonal antibody against PlGF reduced the inflammatory cell infiltration and attenuated the development of atherosclerotic lesions in Apo E KO mice [19].
2.2. Flt-1 and the Soluble Isoform of Flt-1 (sFlt-1)
In 1990, a novel tyrosine kinase receptor, later known as Flt-1, was originally isolated from the human placenta by Shibuya [20]. Unlike Flk-1, the affinity of Flt-1 for its ligand, VEGF, is very high, and is at least 10-fold higher than that for VEGFR-2 [21]. However, the kinase activity of VEGFR-1 is lower, about one-tenth that of VEGFR-2 [20][22]. Flt-1 is primarily expressed on vascular endothelial cells, and it functions mainly to regulate blood vessel angiogenesis, maintenance, and permeability, as well as endothelial cell migration and proliferation [10][23][24]. Its expression on non-vascular cells further supports the non-vascular biological roles of VEGF and PlGF [25][26]. Flt-1 also contributes to the migration and activation of monocytes and macrophages, leading to inflammatory processes and angiogenesis.
In 1993, Kendall reported that a soluble isoform of Flt-1 (sFlt-1) mRNA covers six Ig regions of the full-length Flt-1, with a 31 amino acid–long tail derived from intron 13 [22]. Later investigations reported Flt-1 isoforms that are generated by alternative splicing of exons 14 and 15 [27][28] and by shedding of the Flt-1 extracellular domain [29][30]. sFlt-1 traps PlGF and VEGF-A and acts as a negative regulator of both molecules. These molecular mechanisms indicate that PlGF is pro-atherogenic, while sFlt-1, an intrinsic PlGF antagonist, is anti-atherogenic.
3. PlGF/sFlt-1 Imbalance in CKD
3.1. Circulating PlGF in CKD
Circulating levels of PlGF are normally undetectable, but increased PlGF levels have been described under pathological conditions [31]. In CKD patients, serum levels of PlGF are directly correlated with CKD severity [32] and the left ventricular mass index [33]. It can be found that increasing PlGF levels were associated with an increased risk of both mortality and cardiovascular events, the latter defined as atherosclerotic diseases and heart failure (HF) requiring hospitalization: the risks for patients in the highest quartile (≥19.6 pg/mL) were 3.87- and 8.42-fold higher, respectively, than for those in the lowest quartile (<10.1 pg/mL) [32]. Higher levels of PlGF and greater CKD severity were independently and additively associated with an increased risk of mortality and cardiovascular events (Figure 2).
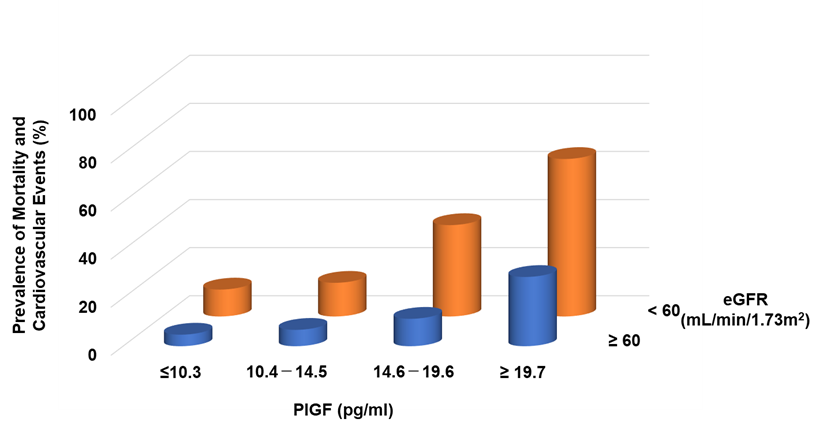

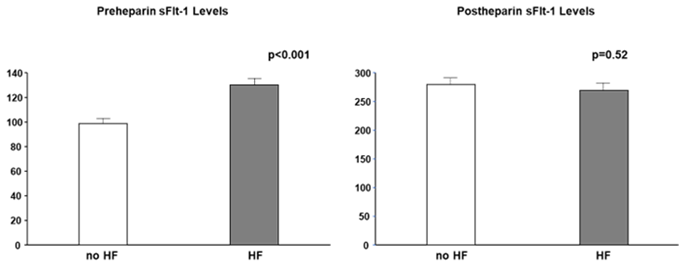

Figure 2. Prevalence of mortality and cardiovascular events in CKD patients. Higher PlGF levels are significantly associated with adverse events.
3.2. PlGF Production in CKD
In a remnant kidney model, PlGF mRNA and protein expression were upregulated not only in the kidneys but also in other organs [34][35][36]. The expression of PlGF in cultured human umbilical arterial endothelial cells was also increased by the addition of human serum from patients with advanced CKD, which also upregulated biomarkers for endothelial injury and oxidative stress. However, treatment with the antioxidant vitamin E attenuated PlGF production [35].
These data suggest that upregulation of PlGF in CKD contributes to the development of arteriosclerotic diseases or HF via augmentation of vascular inflammation and permeability. Activation of mineralocorticoid receptors by aldosterone induces early atherosclerosis and promotes plaque inflammation via increased PlGF secretion from vascular cells [37]. Recently, treatment with a novel selective nonsteroidal mineralocorticoid receptor antagonist, finerenone, reduced the risks of cardiovascular events in patients with diabetic kidney disease [38][39]. This beneficial effect of finerenone in CKD may contribute to the suppression of PlGF/Flt-1 signaling.
4. Role of PlGF/sFlt-1 Imbalance
An in vivo experiment using 5/6-nephrectomized Apo E knockout (KO) mice showed that mRNA and plasma levels of sFlt-1 were decreased but those of PlGF were increased compared with the control Apo E KO mice [35][36]. The CKD model mice had more areas with severe plaque in the thoracoabdominal aorta and aortic root, with massive infiltration of macrophages into the plaques. These atherosclerotic lesions were positively related to plasma levels of indoxyl sulfate and p-cresol [40]. Treatment with AST120, an oral carbon adsorbent of uremic toxins, was inversely related to atherosclerosis in CKD model mice and was also associated with an increase in sFlt-1 production. Additionally, repeated administration of recombinant human sFlt-1 comprising the first three immunoglobulin-like domains of the extracellular region of Flt-1 resulted in significant decreases in plaque area and macrophage infiltration (Figure 3).
Figure 3. Aortic plaque formation in control or 5/6-nephrectomized Apo E KO mice. Plaque formation in the thoracoabdominal aorta was increased in a mouse remnant kidney model but reduced by repeated intraperitoneal injection of sFlt-1.
5. Distribution of sFlt-1
Most studies did not clearly state whether sFlt-1 was measured with or without heparin infusion. Scientists and clinicians must interpret that, in human studies, circulating sFlt-1 levels may be influenced due to heparin infusion since heparin is often preferred in patients with atherosclerosis, heart failure, and preeclampsia to prevent clotting. The heparin loading test is a diagnostic technique of understanding sFlt-1 distribution; pre-heparin sFlt-1 levels represent free sFlt-1 while post-heparin sFlt-1 levels estimate total sFlt-1 (free plus matrix-bound sFlt-1). As mentioned above, CKD presents a mixture of increased free and decreased total sFlt-1 levels. Pre-heparin sFlt-1 levels were significantly higher in HF patients than in non-HF patients, but the two groups were very similar regarding post-heparin levels, which suggests production of sFlt-1 may not be decreased in HF patients without CKD (Figure 4).
Figure 4. Pre- and post-heparin sFlt-1 levels in HF and no HF. Pre-heparin sFlt-1 levels are significantly increased in HF, but post-heparin sFlt-1 levels are similar between HF and no HF. HF is defined by brain natriuretic peptide ≥ 200 pg/mL.
Both persistently high and low levels of sFlt-1 can be considered pathological and can result in arteriosclerosis and cardiac remodeling. These effects are due to the regulation of circulating free sFlt-1, matrix-bound sFlt-1, and total sFlt-1 (free plus matrix-bound sFlt-1) (Figure 5). CKD is characterized by the activation of a number of neurohumoral factors, including the renin–angiotensin–aldosterone system, the sympathetic nervous system, and proinflammatory cytokines such as uremic toxins, all of which lead to endothelial dysfunction [41]. Elevations of free sFlt-1 levels may result not from upregulated sFlt-1 production, but rather from the shifting of matrix-bound sFlt-1 to the circulation as a result of inflammatory endothelial injury in CKD and HF. A recent study showed that sFlt-1 mRNA expression in placental trophoblast cells was unchanged under hypoxic conditions, but sFlt-1 release into the media was significantly greater than under normoxic conditions [42]. A similar phenomenon may occur in ischemic endothelial cells in HF, given that endothelial cells, such as trophoblast cells, are a potential storage site for large amounts of sFlt-1 bound to extracellular heparan sulfate. Elevated sFlt-1 expressions in monocytes isolated from CKD patients and kidney transplantation patients were observed, but there may be other sources increasing free sFlt-1 levels [43][44]. Furthermore, total sFlt-1 is downregulated in the endothelium in CKD, which was confirmed by the finding that post-heparin sFlt-1 levels were decreased in CKD patients. Taken together, the regulation of increased free sFlt-1 levels and decreased total sFlt-1 levels can contribute to the development of atherosclerotic diseases and aggravation of HF in CKD patients.
Figure 5. Circulating sFlt-1, matrix-bound sFlt-1, and total sFlt-1 levels in each model. Heparin infusion releases matrix-bound sFlt-1 into the circulation by displacing the sFlt-1 heparin-binding site from heparin sulfate proteoglycans. In HF, the release of free sFlt-1 from matrix-bound sFlt-1 under hypoxia may increase free sFlt-1 levels. Downregulated endothelial-derived sFlt-1 activates PlGF/Flt-1 signaling in CKD.
This entry is adapted from the peer-reviewed paper 10.3390/ijms232214187
References
- Bikbov, B.; Perico, N.; Remuzzi, G.; on behalf of the GBDGDEG. Disparities in Chronic Kidney Disease Prevalence among Males and Females in 195 Countries: Analysis of the Global Burden of Disease 2016 Study. Nephron 2018, 139, 313–318.
- Adler, A.I.; Stevens, R.J.; Manley, S.E.; Bilous, R.W.; Cull, C.A.; Holman, R.R.; UKPDS Group. Development and progression of nephropathy in type 2 diabetes: The United Kingdom Prospective Diabetes Study (UKPDS 64). Kidney Int. 2003, 63, 225–232.
- Ninomiya, T.; Kiyohara, Y.; Kubo, M.; Tanizaki, Y.; Doi, Y.; Okubo, K.; Wakugawa, Y.; Hata, J.; Oishi, Y.; Shikata, K.; et al. Chronic kidney disease and cardiovascular disease in a general Japanese population: The Hisayama Study. Kidney Int. 2005, 68, 228–236.
- Ronco, C.; Haapio, M.; House, A.A.; Anavekar, N.; Bellomo, R. Cardiorenal syndrome. J. Am. Coll. Cardiol. 2008, 52, 1527–1539.
- Okamoto, R.; Ali, Y.; Hashizume, R.; Suzuki, N.; Ito, M. BNP as a Major Player in the Heart-Kidney Connection. Int. J. Mol. Sci. 2019, 20, 3581.
- Ivey-Miranda, J.B.; Stewart, B.; Cox, Z.L.; McCallum, W.; Maulion, C.; Gleason, O.; Meegan, G.; Amatruda, J.G.; Moreno-Villagomez, J.; Mahoney, D.; et al. FGF-23 (Fibroblast Growth Factor-23) and Cardiorenal Interactions. Circ. Heart Fail. 2021, 14, e008385.7.
- Clementi, A.; Virzi, G.M.; Battaglia, G.G.; Ronco, C. Neurohormonal, Endocrine, and Immune Dysregulation and Inflammation in Cardiorenal Syndrome. Cardiorenal Med. 2019, 9, 265–273.
- Silva-Hucha, S.; Pastor, A.M.; Morcuende, S. Neuroprotective Effect of Vascular Endothelial Growth Factor on Motoneurons of the Oculomotor System. Int. J. Mol. Sci. 2021, 22, 814.
- Alitalo, K.; Tammela, T.; Petrova, T.V. Lymphangiogenesis in development and human disease. Nature 2005, 438, 946–953.
- Autiero, M.; Waltenberger, J.; Communi, D.; Kranz, A.; Moons, L.; Lambrechts, D.; Kroll, J.; Plaisance, S.; De Mol, M.; Bono, F.; et al. Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat. Med. 2003, 9, 936–943.
- Shibuya, M. Vascular endothelial growth factor and its receptor system: Physiological functions in angiogenesis and pathological roles in various diseases. J. Biochem. 2013, 153, 13–19.
- Dabravolski, S.A.; Khotina, V.A.; Omelchenko, A.V.; Kalmykov, V.A.; Orekhov, A.N. The Role of the VEGF Family in Atherosclerosis Development and Its Potential as Treatment Targets. Int. J. Mol. Sci. 2022, 23, 931.
- Maglione, D.; Guerriero, V.; Viglietto, G.; Delli-Bovi, P.; Persico, M.G. Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc. Natl. Acad. Sci. USA 1991, 88, 9267–9271.
- Yonekura, H.; Sakurai, S.; Liu, X.; Migita, H.; Wang, H.; Yamagishi, S.; Nomura, M.; Abedin, M.J.; Unoki, H.; Yamamoto, Y.; et al. Placenta growth factor and vascular endothelial growth factor B and C expression in microvascular endothelial cells and pericytes. Implication in autocrine and paracrine regulation of angiogenesis. J. Biol. Chem. 1999, 274, 35172–35178.
- Persico, M.G.; Vincenti, V.; Di Palma, T. Structure, expression and receptor-binding properties of placenta growth factor (PlGF). Curr. Top. Microbiol. Immunol. 1999, 237, 31–40.
- Iwama, H.; Uemura, S.; Naya, N.; Imagawa, K.; Takemoto, Y.; Asai, O.; Onoue, K.; Okayama, S.; Somekawa, S.; Kida, Y.; et al. Cardiac expression of placental growth factor predicts the improvement of chronic phase left ventricular function in patients with acute myocardial infarction. J. Am. Coll. Cardiol. 2006, 47, 1559–1567.
- Carmeliet, P.; Moons, L.; Luttun, A.; Vincenti, V.; Compernolle, V.; De Mol, M.; Wu, Y.; Bono, F.; Devy, L.; Beck, H.; et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med. 2001, 7, 575–583.
- Khurana, R.; Moons, L.; Shafi, S.; Luttun, A.; Collen, D.; Martin, J.F.; Carmeliet, P.; Zachary, I.C. Placental growth factor promotes atherosclerotic intimal thickening and macrophage accumulation. Circulation 2005, 111, 2828–2836.
- Roncal, C.; Buysschaert, I.; Gerdes, N.; Georgiadou, M.; Ovchinnikova, O.; Fischer, C.; Stassen, J.M.; Moons, L.; Collen, D.; De Bock, K.; et al. Short-term delivery of anti-PlGF antibody delays progression of atherosclerotic plaques to vulnerable lesions. Cardiovasc. Res. 2010, 86, 29–36.
- Shibuya, M.; Yamaguchi, S.; Yamane, A.; Ikeda, T.; Tojo, A.; Matsushime, H.; Sato, M. Nucleotide sequence and expression of a novel human receptor-type tyrosine kinase gene (flt) closely related to the fms family. Oncogene 1990, 5, 519–524.
- Sawano, A.; Takahashi, T.; Yamaguchi, S.; Aonuma, M.; Shibuya, M. Flt-1 but not KDR/Flk-1 tyrosine kinase is a receptor for placenta growth factor, which is related to vascular endothelial growth factor. Cell Growth Differ. 1996, 7, 213–221.
- Kendall, R.L.; Thomas, K.A. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 10705–10709.
- Maharaj, A.S.; Saint-Geniez, M.; Maldonado, A.E.; D’Amore, P.A. Vascular endothelial growth factor localization in the adult. Am. J. Pathol. 2006, 168, 639–648.
- Maharaj, A.S.; D’Amore, P.A. Roles for VEGF in the adult. Microvasc. Res. 2007, 74, 100–113.
- D’Amore, P.A. Vascular endothelial cell growth factor-a: Not just for endothelial cells anymore. Am. J. Pathol. 2007, 171, 14–18.
- Oura, H.; Bertoncini, J.; Velasco, P.; Brown, L.F.; Carmeliet, P.; Detmar, M. A critical role of placental growth factor in the induction of inflammation and edema formation. Blood 2003, 101, 560–567.
- Sela, S.; Itin, A.; Natanson-Yaron, S.; Greenfield, C.; Goldman-Wohl, D.; Yagel, S.; Keshet, E. A novel human-specific soluble vascular endothelial growth factor receptor 1: Cell-type-specific splicing and implications to vascular endothelial growth factor homeostasis and preeclampsia. Circ. Res. 2008, 102, 1566–1574.
- Jebbink, J.; Keijser, R.; Veenboer, G.; van der Post, J.; Ris-Stalpers, C.; Afink, G. Expression of placental FLT1 transcript variants relates to both gestational hypertensive disease and fetal growth. Hypertension 2011, 58, 70–76.
- Rosenberg, V.A.; Buhimschi, I.A.; Lockwood, C.J.; Paidas, M.J.; Dulay, A.T.; Ramma, W.; Abdel-Razeq, S.S.; Zhao, G.; Ahmad, S.; Ahmed, A.; et al. Heparin elevates circulating soluble fms-like tyrosine kinase-1 immunoreactivity in pregnant women receiving anticoagulation therapy. Circulation 2011, 124, 2543–2553.
- Sela, S.; Natanson-Yaron, S.; Zcharia, E.; Vlodavsky, I.; Yagel, S.; Keshet, E. Local retention versus systemic release of soluble VEGF receptor-1 are mediated by heparin-binding and regulated by heparanase. Circ. Res. 2011, 108, 1063–1070.
- Pilarczyk, K.; Sattler, K.J.E.; Galili, O.; Versari, D.; Olson, M.L.; Meyer, F.B.; Zhu, X.Y.; Lerman, L.O.; Lerman, A. Placenta growth factor expression in human atherosclerotic carotid plaques is related to plaque destabilization. Atherosclerosis 2008, 196, 333–340.
- Matsui, M.; Takeda, Y.; Uemura, S.; Matsumoto, T.; Seno, A.; Onoue, K.; Tsushima, H.; Morimoto, K.; Soeda, T.; Okayama, S.; et al. Placental Growth Factor as a Predictor of Cardiovascular Events in Patients with CKD from the NARA-CKD Study. J. Am. Soc. Nephrol. 2015, 26, 2871–2881.
- Peiskerova, M.; Kalousova, M.; Danzig, V.; Mikova, B.; Hodkova, M.; Nemecek, E.; Bani-Hani, A.; Ambroz, D.; Benakova, H.; Linhart, A.; et al. Placental growth factor may predict increased left ventricular mass index in patients with mild to moderate chronic kidney disease--a prospective observational study. BMC Nephrol. 2013, 14, 142.
- Wu, Q.; Du, Y.; Yang, N.; Liang, Y.; Li, Y. Microvasculature change and placenta growth factor expression in the early stage of a rat remnant kidney model. Am. J. Nephrol. 2006, 26, 97–104.
- Matsui, M.; Takeda, Y.; Uemura, S.; Matsumoto, T.; Seno, A.; Onoue, K.; Tsushima, H.; Morimoto, K.; Soeda, T.; Okayama, S.; et al. Suppressed soluble Fms-like tyrosine kinase-1 production aggravates atherosclerosis in chronic kidney disease. Kidney Int. 2014, 85, 393–403.
- Onoue, K.; Uemura, S.; Takeda, Y.; Somekawa, S.; Iwama, H.; Imagawa, K.; Nishida, T.; Morikawa, Y.; Takemoto, Y.; Asai, O.; et al. Reduction of circulating soluble fms-like tyrosine kinase-1 plays a significant role in renal dysfunction-associated aggravation of atherosclerosis. Circulation 2009, 120, 2470–2477.
- McGraw, A.P.; Bagley, J.; Chen, W.S.; Galayda, C.; Nickerson, H.; Armani, A.; Caprio, M.; Carmeliet, P.; Jaffe, I.Z. Aldosterone increases early atherosclerosis and promotes plaque inflammation through a placental growth factor-dependent mechanism. J. Am. Heart Assoc. 2013, 2, e000018.
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229.
- Pitt, B.; Filippatos, G.; Agarwal, R.; Anker, S.D.; Bakris, G.L.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Schloemer, P.; et al. Cardiovascular Events with Finerenone in Kidney Disease and Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 2252–2263.
- Nakada, Y.; Onoue, K.; Nakano, T.; Ishihara, S.; Kumazawa, T.; Nakagawa, H.; Ueda, T.; Nishida, T.; Soeda, T.; Okayama, S.; et al. AST-120, an Oral Carbon Absorbent, Protects against the Progression of Atherosclerosis in a Mouse Chronic Renal Failure Model by Preserving sFlt-1 Expression Levels. Sci. Rep. 2019, 9, 15571.
- Savira, F.; Magaye, R.; Liew, D.; Reid, C.; Kelly, D.J.; Kompa, A.R.; Sangaralingham, S.J.; Burnett, J.C.; Kaye, D., Jr.; Wang, B.H. Cardiorenal syndrome: Multi-organ dysfunction involving the heart, kidney and vasculature. Br. J. Pharmacol. 2020, 177, 2906–2922.
- Eddy, A.C.; Chapman, H.; George, E.M. Heparanase regulation of sFLT-1 release in trophoblasts in vitro. Placenta 2019, 85, 63–68.
- Wewers, T.M.; Schulz, A.; Nolte, I.; Pavenstädt, H.; Brand, M.; Di Marco, G.S. Circulating Soluble Fms-like Tyrosine Kinase in Renal Diseases Other than Preeclampsia. J. Am. Soc. Nephrol. 2021, 32, 1853–1863.
- Wewers, T.M.; Mayer, A.B.; Pfleiderer, A.; Beul, K.; Schmidt, R.; Heitplatz, B.; Van Marck, V.; Nolte, I.; Pavenstädt, H.; Reuter, S.; et al. Increased soluble fms-like tyrosine kinase 1 after ischemia reperfusion contributes to adverse clinical outcomes following kidney transplantation. Kidney Int. 2019, 95, 1091–1102.
This entry is offline, you can click here to edit this entry!