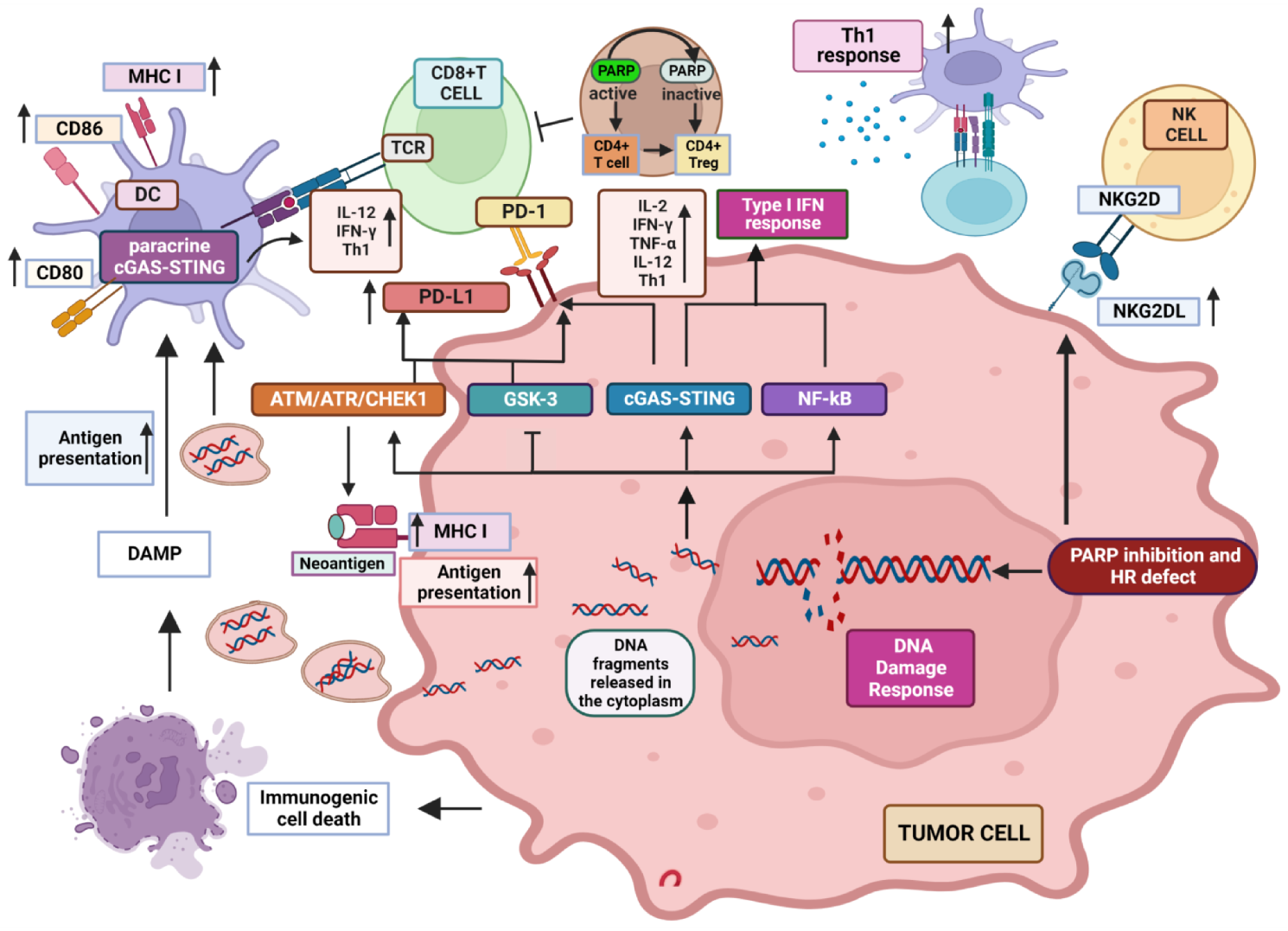
Poly (ADP-ribose) polymerase (PARP) inhibitors (PARPi) induce cytotoxic effects as single agents in tumors characterized by defective repair of DNA double-strand breaks deriving from BRCA1/2 mutations or other abnormalities in genes associated with homologous recombination. Preclinical studies have shown that PARPi-induced DNA damage may affect the tumor immune microenvironment and immune-mediated anti-tumor response through several mechanisms. In particular, increased DNA damage has been shown to induce the activation of type I interferon pathway and up-regulation of PD-L1 expression in cancer cells, which can both enhance sensitivity to Immune Checkpoint Inhibitors (ICIs).
- PARP inhibitor
- BRCA
- DNA damage response
- immunotherapy
1. Introduction
2. Influence of PARP Inhibition on Immune Response to Tumors
2.1. Well Established Effects of PARP Inhibition on the Different Features of TME
2.2. PARP-1 Inhibition Favors NKG2D Activity
This entry is adapted from the peer-reviewed paper 10.3390/cancers14225633
References
- Vyas, S.; Chesarone-Cataldo, M.; Todorova, T.; Huang, Y.H.; Chang, P. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat. Commun. 2013, 4, 2240.
- Vyas, S.; Matic, I.; Uchima, L.; Rood, J.; Zaja, R.; Hay, R.T.; Ahel, I.; Chang, P. Family-wide analysis of poly (ADP-ribose) polymerase activity. Nat. Commun. 2014, 5, 4426.
- Fisher, A.E.; Hochegger, H.; Takeda, S.; Caldecott, K.W. Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydro-lase. Mol. Cell. Biol. 2007, 27, 5597–5605.
- Alemasova, E.E.; Larvik, O.I. Poly(ADP-ribosyl)ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019, 47, 3811–3827.
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182.
- Sugimura, K.; Takebayashi, S.; Taguchi, H.; Takeda, S.; Okumura, K. PARP-1 ensures regulation of replication fork progression by homologous recombination on damaged DNA. J. Cell Biol. 2008, 183, 1203–1212.
- Couto, C.A.; Wang, H.Y.; Green, J.C.; Kiely, R.; Siddaway, R.; Borer, C.; Pears, C.J.; Lakin, N.D. PARP regulates nonhomologous end joining through retention of Ku at double-strand breaks. J. Cell Biol. 2011, 194, 367–375.
- De Lorenzo, S.B.; Patel, A.G.; Hurley, R.M.; Kaufmann, S.H. The Elephant and the Blind Men: Making Sense of PARP Inhibitors in Homologous Recombination Deficient Tumor Cells. Front. Oncol. 2013, 3, 228.
- Hanzlikova, H.; Kalasova, I.; Demin, A.A.; Pennicott, L.E.; Cihlarova, Z.; Caldecott, K.W. The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol. Cell. 2018, 71, 319–331.
- Wang, Y.; Luo, W.; Wang, Y. PARP-1 and its associated nucleases in DNA damage response. DNA Rep. 2019, 81, 102651.
- Krishnakumar, R.; Kraus, W.L. The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell. 2010, 39, 8–24.
- Weaver, A.N.; Yang, E.S. Beyond DNA Repair: Additional Functions of PARP-1 in Cancer. Front. Oncol. 2013, 3, 290.
- Langelier, M.F.; Riccio, A.A.; Pascal, J.M. PARP-2 and PARP-3 are selectively activated by 5’ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res. 2014, 42, 7762–7775.
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination-Related Gene Mutations Across Multiple Cancer Types. J. Clin. Oncol. 2018, 2, 1–13.
- Helleday, T.; Bryant, H.E.; Schultz, N. Poly(ADP-ribose) polymerase (PARP-1) in homologous recombination and as a target for cancer therapy. Cell Cycle 2005, 4, 1176–1178.
- Hu, Y.; Petit, S.A.; Ficarro, S.B.; Toomire, K.J.; Xie, A.; Lim, E.; Cao, S.A.; Park, E.; Eck, M.J.; Scully, R.; et al. PARP1-driven poly-ADP-ribosylation regulates BRCA1 function in homologous recombination-mediated DNA repair. Cancer Discov. 2014, 4, 1430–1447.
- De Vos, M.; Schreiber, V.; Dantzer, F. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem. Pharmacol. 2012, 84, 137–146.
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921.
- King, M.C.; Marks, J.H.; Mandell, J.B. New York Breast Cancer Study Group. Breast and ovarian cancer risk due to in-herited mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646.
- Cavanagh, H.; Rogers, K.M. The role of BRCA1 and BRCA2 mutations in prostate, pancreatic and stomach cancers. Her. Cancer Clin. Pract. 2015, 13, 16.
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228.
- Mizukami, K.; Iwasaki, Y.; Kawakami, E.; Hirata, M.; Kamatani, Y.; Matsuda, K.; Endo, M.; Sugano, K.; Yoshida, T.; Murakami, Y.; et al. Genetic characterization of pancreatic cancer patients and prediction of carrier status of germline pathogenic variants in cancer-predisposing genes. EBioMed 2020, 60, 103033.
- Liao, H.; Ji, F.; Helleday, T.; Ying, S. Mechanisms for stalled replication fork stabiliza-tion: New targets for synthetic lethality strategies in cancer treatments. EMBO Rep. 2018, 19, e46263.
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917.
- Patel, A.G.; Sarkaria, J.N.; Kaufmann, S.H. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3406–3411.
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599.
- Murai, J.; Huang, S.Y.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443.
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps17.
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533.
- Faraoni, I.; Graziani, G. Role of BRCA Mutations in Cancer Treatment with Poly(ADP-ribose) Polymerase (PARP) Inhibitors. Cancers 2018, 10, 487.
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763.
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505.
- Wang, H.; Wu, M.; Liu, H.; Zhou, H.; Zhao, Y.; Geng, Y.; Jiang, B.; Zhang, K.; Zhang, B.; Han, Z.; et al. Comparison of the Efficacy and Safety of PARP Inhibitors as a Monotherapy for Platinum-Sensitive Recurrent Ovarian Cancer: A Network Meta-Analysis. Front. Oncol. 2021, 11, 785102.
- Shen, Y.; Aoyagi-Scharber, M.; Wang, B. Trapping Poly(ADP-Ribose) Polymerase. J. Pharmacol. Exp. Ther. 2015, 353, 44657.
- Boussios, S.; Abson, C.; Moschetta, M.; Rassy, E.; Karathanasi, A.; Bhat, T.; Ghumman, F.; Sheriff, M.; Pavlidis, N. Poly (ADP-Ribose) Polymerase Inhibitors: Talazoparib in Ovarian Cancer and Beyond. Drugs R D 2020, 20, 55–73.
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861.
- Mullard, A. European regulators approve first PARP inhibitor. Nat. Rev. Drug Discov. 2014, 13, 877.
- Taylor, A.M.; Chan, D.; Tio, M.; Patil, S.M.; Traina, T.A.; Robson, M.E.; Khasraw, M. PARP (Poly ADP-Ribose Polymer-ase) inhibitors for locally advanced or metastatic breast cancer. Cochrane Database Syst. Rev. 2021, 4, CD011395.
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell. Dev. Biol. 2020, 8, 564601.
- Tew, W.P.; Lacchetti, C.; Ellis, A.; Maxian, K.; Banerjee, S.; Bookman, M.; Jones, M.B.; Lee, J.M.; Lheureux, S.; Liu, J.F.; et al. PARP Inhibitors in the Management of Ovarian Cancer: ASCO Guideline. J. Clin. Oncol. 2020, 38, 3468–3493.
- Tattersall, A.; Ryan, N.; Wiggans, A.J.; Rogozińska, E.; Morrison, J. Poly(ADP-ribose) polymerase (PARP) inhibitors for the treatment of ovarian cancer. Cochrane Database Syst. Rev. 2022, 2, CD007929.
- Banerjee, S.; Moore, K.N.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; et al. Maintenance olaparib for patients with newly diagnosed advanced ovarian cancer and a BRCA mutation (SOLO1/GOG 3004): 5-year follow-up of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2021, 22, 1721–1731.
- Zam, W.; Ali, L. Immune Checkpoint Inhibitors in the Treatment of Cancer. Curr. Rev. Clin. Exp. Pharmacol. 2022, 17, 103–113.
- Schoenfeld, A.J.; Hellmann, M.D. Acquired Resistance to Immune Checkpoint Inhibitors. Cancer Cell. 2020, 37, 443–455.
- Miao, D.; Margolis, C.A.; Vokes, N.I.; Liu, D.; Taylor-Weiner, A.; Wankowicz, S.M.; Adeegbe, D.; Keliher, D.; Schilling, B.; Tracy, A.; et al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat. Genet. 2018, 50, 1271–1281.
- Yi, M.; Qin, S.; Zhao, W.; Yu, S.; Chu, Q.; Wu, K. The role of neoantigen in immune checkpoint blockade therapy. Exp. Hematol. Oncol. 2018, 7, 28.
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10.
- Zhang, Z.; Lu, M.; Qin, Y.; Gao, W.; Tao, L.; Su, W.; Zhong, J. Neoantigen: A New Breakthrough in Tumor Immunotherapy. Front. Immunol. 2021, 12, 672356.
- Kim, I.S.; Gao, Y.; Welte, T.; Wang, H.; Liu, J.; Janghorban, M.; Sheng, K.; Niu, Y.; Goldstein, A.; Zhao, N.; et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat. Cell. Biol. 2019, 21, 1113–1126.
- Martinez, A.; Delord, J.P.; Ayyoub, M.; Devaud, C. Preclinical and Clinical Immunotherapeutic Strategies in Epithelial Ovarian Cancer. Cancers 2020, 12, 1761.
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301.
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819.
- Mendes-Pereira, A.M.; Martin, S.A.; Brough, R.; McCarthy, A.; Taylor, J.R.; Kim, J.S.; Waldman, T.; Lord, C.J.; Ashworth, A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 2009, 1, 315–322.
- Cerrato, A.; Morra, F.; Celetti, A. Use of poly ADP-ribose polymerase inhibitors in cancer cells bearing DDR defects: The rationale for their inclusion in the clinic. J. Exp. Clin. Cancer Res. 2016, 35, 179.
- Keung, M.; Wu, Y.; Vadgama, J.V. PARP Inhibitors as a Therapeutic Agent for Homologous Recombination Deficiency in Breast Cancers. J. Clin. Med. 2019, 8, 435.
- Germani, A.; Petrucci, S.; De Marchis, L.; Libi, F.; Savio, C.; Amanti, C.; Bonifacino, A.; Campanella, B.; Capalbo, C.; Lombardi, A.; et al. Beyond BRCA1 and BRCA2: Deleterious Variants in DNA Repair Pathway Genes in Italian Families with Breast/Ovarian and Pancreatic Cancers. J. Clin. Med. 2020, 9, 3003.
- Dalmasso, B.; Puccini, A.; Catalano, F.; Borea, R.; Iaia, M.L.; Bruno, W.; Fornarini, G.; Sciallero, S.; Rebuzzi, S.E.; Ghiorzo, P. Beyond BRCA: The Emerging Significance of DNA Damage Response and Personalized Treatment in Pancreatic and Prostate Cancer Patients. Int. J. Mol. Sci. 2022, 23, 4709.
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520.
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016, 7, 13587–13598.
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov. 2017, 7, 264–276.
- Pfeifer, G.P. Environmental exposures and mutational patterns of cancer genomes. Genome Med. 2010, 2, 54.
- Hussein, Y.R.; Weigelt, B.; Levine, D.A.; Schoolmeester, J.K.; Dao, L.N.; Balzer, B.L.; Liles, G.; Karlan, B.; Köbel, M.; Lee, C.H.; et al. Clinicopathological analysis of endometrial carcinomas harboring somatic POLE exonuclease domain mutations. Modern Pathol. 2015, 28, 505–514.
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51.
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61.
- Xiao, Y.; Freeman, G.J. The microsatellite instable subset of colorectal cancer is a particularly good candidate for checkpoint blockade immunotherapy. Cancer Discov. 2015, 5, 16–18.
- Samstein, R.M.; Riaz, N. The DNA damage response in immunotherapy and radiation. Adv. Rad. Oncol. 2018, 3, 527–533.
- Vidotto, T.; Nersesian, S.; Graham, C.; Siemens, D.R.; Koti, M. DNA damage repair gene mutations and their association with tumor immune regulatory gene expression in muscle invasive bladder cancer subtypes. J. Immunother. Cancer 2019, 7, 148.
- Zou, X.L.; Li, X.B.; Ke, H.; Zhang, G.Y.; Tang, Q.; Yuan, J.; Zhou, C.J.; Zhang, J.L.; Zhang, R.; Chen, W.Y. Prognostic Value of Neoantigen Load in Immune Checkpoint Inhibitor Therapy for Cancer. Front. Immunol. 2021, 12, 689076.
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299.
- Jiang, M.; Jia, K.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; He, Y.; Zhou, C. Alterations of DNA damage response pathway: Biomarker and therapeutic strategy for cancer immunotherapy. Acta Pharm. Sin. B 2021, 11, 2983–2994.
- Tang, M.L.; Khan, M.K.; Croxford, J.L.; Tan, K.W.; Angeli, V.; Gasser, S. The DNA damage response induces antigen presenting cell-like functions in fibroblasts. Eur. J. Immunol. 2014, 44, 1108–1118.
- Seyedin, S.N.; Hasibuzzaman, M.M.; Pham, V.; Petronek, M.S.; Callaghan, C.; Kalen, A.L.; Mapuskar, K.A.; Mott, S.L.; Spitz, D.R.; Allen, B.G.; et al. Combination Therapy with Radiation and PARP Inhibition Enhances Responsiveness to Anti-PD-1 Therapy in Colorectal Tumor Models. Int. Ernational J. Rad. Oncol. Biol. Phys. 2020, 108, 81–92.
- Brown, J.S.; Sundar, R.; Lopez, J. Combining DNA damaging therapeutics with immunotherapy: More haste, less speed. Br. J. Cancer 2018, 118, 312–324.
- Tanaka, Y.; Chen, Z.J. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal 2012, 5, ra20.
- Ho, S.S.; Zhang, W.Y.; Tan, N.Y.; Khatoo, M.; Suter, M.A.; Tripathi, S.; Cheung, F.S.; Lim, W.K.; Tan, P.H.; Ngeow, J.; et al. The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity 2016, 44, 1177–1189.
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472.
- Ishikawa, H.; Barber, G. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678.
- Barber, G. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770.
- Corrales, L.; McWhirter, S.M.; Dubensky, T.W., Jr.; Gajewski, T.F. The host STING pathway at the interface of cancer and immunity. J. Clin. Investig. 2016, 126, 2404–2411.
- Jing, W.; McAllister, D.; Vonderhaar, E.P.; Palen, K.; Riese, M.J.; Gershan, J.; Johnson, B.D.; Dwinell, M.B. STING agonist inflames the pancreatic cancer immune microenvironment and reduces tumor burden in mouse models. J. Immunother. Cancer 2019, 7, 115.
- Wang, Z.; Sun, K.; Xiao, Y.; Feng, B.; Mikule, K.; Ma, X.; Feng, N.; Vellano, C.P.; Federico, L.; Marszalek, J.R.; et al. Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Sci. Rep. 2019, 9, 1853.
- Neufeldt, C.J.; Cerikan, B.; Cortese, M.; Frankish, J.; Lee, J.Y.; Plociennikowska, A.; Heigwer, F.; Prasad, V.; Joecks, S.; Burkart, S.S.; et al. SARS-CoV-2 infection induces a pro-inflammatory cytokine response through cGAS-STING and NF-κB. Commun. Biol. 2022, 5, 45.
- Garris, C.S.; Arlauckas, S.P.; Kohler, R.H.; Trefny, M.P.; Garren, S.; Piot, C.; Engblom, C.; Pfirschke, C.; Siwicki, M.; Gungabeesoon, J.; et al. Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-γ and IL-12. Immunity 2018, 49, 1148–1161.e7.
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Sign. Trans. Target. Ther. 2017, 2, 17023.
- Abe, T.; Barber, G.N. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J. Virol. 2014, 88, 5328–5341.
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-phase-Specific DNA Damage in Breast Cancer. J. Nat. Cancer Inst. 2016, 109, djw199.
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8+ T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737.
- Lee, E.K.; Konstantinopoulos, P.A. PARP inhibition and immune modulation: Scientific rationale and perspectives for the treatment of gynecologic cancers. Ther. Adv. Med. Oncol. 2020, 12, 1758835920944116.
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569.
- Yélamos, J.; Moreno-Lama, L.; Jimeno, J.; Ali, S.O. Immunomodulatory Roles of PARP-1 and PARP-2: Impact on PARP-Centered Cancer Therapies. Cancers 2020, 12, 392.
- Tang, T.; Cheng, X.; Truong, B.; Sun, L.; Yang, X.; Wang, H. Molecular basis and therapeutic implications of CD40/CD40L immune checkpoint. Pharmacol. Ther. 2021, 219, 107709.
- Zhao, Y.; Lee, C.K.; Lin, C.H.; Gassen, R.B.; Xu, X.; Huang, Z.; Xiao, C.; Bonorino, C.; Lu, L.F.; Bui, J.D.; et al. PD-L1:CD80 Cis-Heterodimer Triggers the Co-stimulatory Receptor CD28 While Repressing the Inhibitory PD-1 and CTLA-4 Pathways. Immunity 2019, 51, 1059–1073.
- Ishii, H.; Azuma, K.; Kawahara, A.; Yamada, K.; Imamura, Y.; Tokito, T.; Kinoshita, T.; Kage, M.; Hoshino, T. Significance of programmed cell death-ligand 1 expression and its association with survival in patients with small cell lung cancer. J. Thor. Oncol. 2015, 10, 426–430.
- Paglialunga, L.; Salih, Z.; Ricciuti, B.; Califano, R. Immune checkpoint blockade in small cell lung cancer: Is there a light at the end of the tunnel? ESMO 2016, 1, e000022.
- Byers, L.A.; Wang, J.; Nilsson, M.B.; Fujimoto, J.; Saintigny, P.; Yordy, J.; Giri, U.; Peyton, M.; Fan, Y.H.; Diao, L.; et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2012, 2, 798–811.
- Sen, T.; Rodriguez, B.L.; Chen, L.; Corte, C.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661.
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. 2019, 19, 133–150.
- Huang, J.; Wang, L.; Cong, Z.; Amoozgar, Z.; Kiner, E.; Xing, D.; Orsulic, S.; Matulonis, U.; Goldberg, M.S. The PARP1 inhibitor BMN 673 exhibits immunoregulatory effects in a Brca1(-/-) murine model of ovarian cancer. Biochem. Biophys. Res. Comm. 2015, 463, 551–556.
- Staniszewska, A.D.; Armenia, J.; King, M.; Michaloglou, C.; Reddy, A.; Singh, M.; San Martin, M.; Prickett, L.; Wilson, Z.; Proia, T.; et al. PARP inhibition is a modulator of anti-tumor immune response in BRCA-deficient tumors. Oncoimmunology 2022, 11, 2083755.
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319.
- Iurescia, S.; Fioretti, D.; Rinaldi, M. Targeting Cytosolic Nucleic Acid-Sensing Pathways for Cancer Immunotherapies. Front. Immunol. 2018, 9, 711.
- Schüler, T.; Qin, Z.; Ibe, S.; Noben-Trauth, N.; Blumenstein, T. T helper cell type 1-associated and cytotoxic T lymphocyte-mediated tumor immunity is impaired in interleukin 4-deficient mice. J. Exp. Med. 1999, 189, 803–810.
- Shen, J.; Xiao, Z.; Zhao, Q.; Li, M.; Wu, X.; Zhang, L.; Hu, W.; Cho, C.H. Anti-cancer therapy with TNFα and IFNγ: A comprehensive review. Cell Prolif. 2018, 51, e12441.
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e12.
- Saenz, L.; Lozano, J.J.; Valdor, R.; Baroja-Mazo, A.; Ramirez, P.; Parrilla, P.; Aparicio, P.; Sumoy, L.; Yélamos, J. Transcriptional regulation by poly(ADP-ribose) polymerase-1 during T cell activation. BMC Genom. 2008, 9, 171.
- Datta, R.; Naura, A.S.; Zerfaoui, M.; Errami, Y.; Oumouna, M.; Kim, H.; Ju, J.; Ronchi, V.P.; Haas, A.L.; Boulares, A.H. PARP-1 deficiency blocks IL-5 expression through calpain-dependent degradation of STAT-6 in a murine asthma model. Allergy 2011, 66, 853–861.
- Ghonim, M.A.; Pyakurel, K.; Ibba, S.V.; Al-Khami, A.A.; Wang, J.; Rodriguez, P.; Rady, H.F.; El-Bahrawy, A.H.; Lammi, M.R.; Mansy, M.S.; et al. PARP inhibition by olaparib or gene knockout blocks asthma-like manifestation in mice by modulating CD4(+) T cell function. J. Transl. Med. 2015, 13, 225.
- Zhu, J.; Yamane, H.; Cote-Sierra, J.; Guo, L.; Paul, W.E. GATA-3 promotes Th2 responses through three different mechanisms: Induction of Th2 cytokine production, selective growth of Th2 cells and inhibition of Th1 cell-specific factors. Cell Res. 2005, 16, 3–10.
- Zong, W.; Gong, Y.; Sun, W.; Li, T.; Wang, Z.Q. PARP1: Liaison of Chromatin Remodeling and Transcription. Cancers 2022, 14, 4162.
- Ansel, K.M.; Djuretic, I.; Tanasa, B.; Rao, A. Regulation of Th2 differentiation and Il4 locus accessibility. Ann. Rev. Immunol. 2006, 24, 607–656.
- Zhang, P.; Maruyama, T.; Konkel, J.E.; Abbatiello, B.; Zamarron, B.; Wang, Z.Q.; Chen, W. PARP-1 controls immunosuppressive function of regulatory T cells by destabilizing Foxp3. PLoS ONE 2013, 8, e71590.
- Luo, X.; Nie, J.; Wang, S.; Chen, Z.; Chen, W.; Li, D.; Hu, H.; Li, B. Poly(ADP-ribosyl)ation of FOXP3 Protein Mediated by PARP-1 Protein Regulates the Function of Regulatory T Cells. J. Biol. Chem. 2015, 290, 28675–28682.
- Nasta, F.; Laudisi, F.; Sambucci, M.; Rosado, M.M.; Pioli, C. Increased Foxp3+ regulatory T cells in poly(ADP-Ribose) polymerase-1 deficiency. J. Immunol. 2010, 184, 3470–3477.
- Wang, Y.; Tong, Z.; Zhang, W.; Zhang, W.; Buzdin, A.; Mu, X.; Yan, Q.; Zhao, X.; Chang, H.H.; Duhon, M.; et al. FDA-Approved and Emerging Next Generation Predictive Biomarkers for Immune Checkpoint Inhibitors in Cancer Patients. Front. Oncol. 2021, 11, 683419.
- Yum, S.; Li, M.; Fang, Y.; Chen, Z.J. TBK1 recruitment to STING activates both IRF3 and NF-κB that mediate immune defense against tumors and viral infections. Proc. Natl. Acad. Sci. USA 2021, 118, e2100225118.
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751.
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expres-sion and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720.
- Li, C.W.; Lim, S.O.; Xia, W.; Lee, H.H.; Chan, L.C.; Kuo, C.W.; Khoo, K.H.; Chang, S.S.; Cha, J.H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7, 12632.
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742.
- Cohen, P.; Frame, S. The renaissance of GSK3. Nat. Rev. 2001, 2, 769–776.
- Yélamos, J.; Monreal, Y.; Saenz, L.; Aguado, E.; Schreiber, V.; Mota, R.; Fuente, T.; Minguela, A.; Parrilla, P.; de Murcia, G.; et al. PARP-2 deficiency affects the survival of CD4+CD8+ double-positive thymocytes. EMBO J. 2006, 25, 4350–4360.
- Ding, L.; Kim, H.J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP inhibition elicits STING-dependent antitumor immunity in brca1-deficient ovarian cancer. Cell Rep. 2018, 25, 2972–2980.e5.
- Samstein, R.M.; Krishna, C.; Ma, X.; Pei, X.; Lee, K.W.; Makarov, V.; Kuo, F.; Chung, J.; Srivastava, R.M.; Purohit, T.A.; et al. Mutations in BRCA1 and BRCA2 differentially affect the tumor microenvironment and response to checkpoint blockade immunotherapy. Nat. Cancer 2021, 1, 1188–1203.
- Lan, T.; Chen, L.; Wei, X. Inflammatory Cytokines in Cancer: Comprehensive Understanding and Clinical Progress in Gene Therapy. Cells 2021, 10, 100.
- Aldinucci, D.; Colombatti, A. The inflammatory chemokine CCL5 and cancer progression. Mediat. Inflamm. 2014, 2014, 292376.
- Khalid, A.; Wolfram, J.; Ferrari, I.; Mu, C.; Mai, J.; Yang, Z.; Zhao, Y.; Ferrari, M.; Ma, X.; Shen, H. Recent Advances in Discovering the Role of CCL5 in Metastatic Breast Cancer. Mini Rev. Med. Chem. 2015, 15, 1063–1072.
- Aldinucci, D.; Borghese, C.; Casagrande, N. The CCL5/CCR5 Axis in Cancer Progression. Cancers 2020, 12, 1765.
- Li, X.; Fang, T.; Xu, S.; Jin, P.; Zhou, D.; Wang, Z.; Li, H.; Yang, Z.; Chen, G.; Zheng, X.; et al. PARP inhibitors promote stromal fibroblast activation by enhancing CCL5 autocrine signaling in ovarian cancer. NPJ Precis. Oncol. 2021, 5, 49.
- Huang, C.Y.; Fong, Y.C.; Lee, C.Y.; Chen, M.Y.; Tsai, H.C.; Hsu, H.C.; Tang, C.H. CCL5 increases lung cancer migration via PI3K, Akt and NF-kappaB pathways. Biochem. Pharmacol. 2009, 77, 794–803.
- Kim, J.E.; Kim, H.S.; Shin, Y.J.; Lee, C.S.; Won, C.; Lee, S.A.; Lee, J.W.; Kim, Y.; Kang, J.S.; Ye, S.K.; et al. LYR71, a derivative of trimeric resveratrol, inhibits tumorigenesis by blocking STAT3-mediated matrix metalloproteinase 9 expression. Exp. Mol. Med. 2008, 40, 514–522.
- Kato, T.; Fujita, Y.; Nakane, K.; Mizutani, K.; Terazawa, R.; Ehara, H.; Kanimoto, Y.; Kojima, T.; Nozawa, Y.; Deguchi, T.; et al. CCR1/CCL5 interaction promotes invasion of taxane-resistant PC3 prostate cancer cells by increasing secretion of MMPs 2/9 and by activating ERK and Rac signaling. Cytokine 2013, 64, 251–257.
- Li, F.; Wu, X.; Fu, X.; Liu, J.; Song, W.; Xiao, G.G.; Lu, A.; Zhang, G. Poly (ADP-ribose) polymerase 1 (PARP1) inhibition promotes pulmonary metastasis of osteosarcoma by boosting ezrin phosphorylation. Int. J. Biol. Sci. 2022, 18, 1238–1253.
- Clucas, J.; Valderrama, F. ERM proteins in cancer progression. J. Cell Sci. 2014, 127, 267–275.
- Moilanen, J.; Lassus, H.; Leminen, A.; Vaheri, A.; Bützow, R.; Carpén, O. Ezrin immunoreactivity in relation to survival in serous ovari-an carcinoma patients. Gynecol. Oncol. 2003, 90, 273–281.
- Horwitz, V.; Davidson, B.; Stern, D.; Tropé, C.G.; Tavor Re’em, T.; Reich, R. Ezrin Is Associated with Disease Progression in Ovarian Carcinoma. PLoS ONE 2016, 11, e0162502.
- Houchins, J.P.; Yabe, T.; McSherry, C.; Bach, F.H. DNA sequence analysis of NKG2, a family of related cDNA clones en-coding type II integral membrane proteins on human natural killer cells. J. Exp. Med. 1991, 173, 1017–1020.
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999, 285, 727–729.
- Jamieson, A.M.; Diefenbach, A.; McMahon, C.W.; Xiong, N.; Carlyle, J.R.; Raulet, D.H. The role of the NKG2D immuno-receptor in immune cell activation and natural killing. Immunity 2002, 17, 19–29.
- Fuertes, M.B.; Domaica, C.I.; Zwirner, N.W. Leveraging NKG2D Ligands in Immuno-Oncology. Front. Immunol. 2021, 12, 713158.
- Diefenbach, A.; Jamieson, A.M.; Liu, S.D.; Shastri, N.; Raulet, D.H. Ligands for the murine NKG2D receptor: Expression by tumor cells and activation of NK cells and macrophages. Nat. Immunol. 2000, 1, 119–126.
- Maasho, K.; Opoku-Anane, J.; Marusina, A.I.; Coligan, J.E.; Borrego, F. NKG2D is a costimulatory receptor for human naive CD8+ T cells. J. Immunol. 2005, 174, 4480–4484.
- Markiewicz, M.A.; Carayannopoulos, L.N.; Naidenko, O.V.; Matsui, K.; Burack, W.R.; Wise, E.L.; Fremont, D.H.; Allen, P.M.; Yokoyama, W.M.; Colonna, M.; et al. Costimulation through NKG2D enhances murine CD8+ CTL function: Similarities and differences between NKG2D and CD28 costimulation. J. Immunol. 2005, 175, 2825–2833.
- Prajapati, K.; Perez, C.; Rojas, L.; Burke, B.; Guevara-Patino, J.A. Functions of NKG2D in CD8+ T cells: An opportunity for immunotherapy. Cell. Mol. Immunol. 2018, 15, 470–479.
- Rajasekaran, K.; Xiong, V.; Fong, L.; Gorski, J.; Malarkannan, S. Functional dichotomy between NKG2D and CD28-mediated co-stimulation in human CD8+ T cells. PLoS ONE 2010, 5, e12635.
- Lee, K.P.; Taylor, C.; Petryniak, B.; Turka, L.A.; June, C.H.; Thompson, C.B. The genomic organization of the CD28 gene. Implications for the regulation of CD28 mRNA expression and heterogeneity. J. Immunol. 1990, 145, 344–352.
- Plunkett, F.J.; Franzese, O.; Finney, H.M.; Fletcher, J.M.; Belaramani, L.L.; Salmon, M.; Dokal, I.; Webster, D.; Lawson, A.D.; Akbar, A. The loss of telomerase activity in highly differentiated CD8+CD28-CD27- T cells is associated with decreased Akt (Ser473) phosphorylation. J. Immunol. 2007, 178, 7710–7719.
- Zingoni, A.; Molfetta, R.; Fionda, C.; Soriani, A.; Paolini, R.; Cippitelli, M.; Cerboni, C.; Santoni, A. NKG2D and Its Ligands: “One for All, All for One”. Front. Immunol. 2018, 9, 476.
- Cerwenka, A.; Bakker, A.B.; McClanahan, T.; Wagner, J.; Wu, J.; Phillips, J.H.; Lanier, L.L. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity 2000, 12, 721–727.
- O’Callaghan, C.A.; Cerwenka, A.; Willcox, B.E.; Lanier, L.L.; Bjorkman, P.J. Molecular competition for NKG2D: H60 and RAE1 compete unequally for NKG2D with dominance of H60. Immunity 2001, 15, 201–211.
- Champsaur, M.; Lanier, L.L. Effect of NKG2D ligand expression on host immune responses. Immunol. Rev. 2010, 235, 267–285.
- Kuylenstierna, C.; Björkström, N.K.; Andersson, S.K.; Sahlström, P.; Bosnjak, L.; Paquin-Proulx, D.; Malmberg, K.J.; Ljunggren, H.G.; Moll, M.; Sandberg, J.K. NKG2D performs two functions in invariant NKT cells: Direct TCR-independent activation of NK-like cytolysis and co-stimulation of activation by CD1d. Eur. J. Immunol. 2011, 41, 1913–1923.
- Lanier, L.L. NKG2D receptor and its ligands in host defense. Cancer Immunol. Res. 2015, 3, 575–582.
- Nowbakht, P.; Ionescu, M.C.; Rohner, A.; Kalberer, C.P.; Rossy, E.; Mori, L.; Cosman, D.; De Libero, G.; Wodnar-Filipowicz, A. Ligands for natural killer cell-activating receptors are expressed upon the maturation of normal myelo-monocytic cells but at low levels in acute myeloid leukemias. Blood 2005, 105, 3615–3622.
- Hilpert, J.; Grosse-Hovest, L.; Grünebach, F.; Buechele, C.; Nuebling, T.; Raum, T.; Steinle, A.; Salih, H.R. Comprehensive analysis of NKG2D ligand expression and release in leukemia: Implications for NKG2D-mediated NK cell responses. J. Immunol. 2012, 189, 1360–1371.
- Saito, Y.; Kitamura, H.; Hijikata, A.; Tomizawa-Murasawa, M.; Tanaka, S.; Takagi, S.; Uchida, N.; Suzuki, N.; Sone, A.; Najima, Y.; et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci. Transl. Med. 2010, 2, 17ra9.
- Ng, S.W.; Mitchell, A.; Kennedy, J.A.; Chen, W.C.; McLeod, J.; Ibrahimov, N.; Arruda, A.; Popescu, A.; Gupta, V.; Shimmer, A.D.; et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016, 540, 433–437.
- Fauriat, C.; Just-Landi, S.; Mallet, F.; Arnoulet, C.; Sainty, D.; Olive, D.; Costello, R.T. Deficient expression of NCR in NK cells from acute myeloid leukemia: Evolution during leukemia treatment and impact of leukemia cells in NCRdull phenotype induction. Blood 2007, 109, 323–330.
- Paczulla, A.M.; Rothfelder, K.; Raffel, S.; Konantz, M.; Steinbacher, J.; Wang, H.; Tandler, C.; Mbarga, M.; Schaefer, T.; Falcone, M.; et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 2019, 572, 254–259.
- Vairy, S.; Garcia, J.L.; Teira, P.; Bittencourt, H. CTL019 (tisagenlecleucel): CAR-T therapy for relapsed and refractory B-cell acute lymphoblastic leukemia. Drug Des.Dev. Ther. 2018, 12, 3885–3898.
- Nikiforow, S.; Murad, J.; Daley, H.; Negre, H.; Reder, J.; Sentman, C.L.; Lehmann, F.; Snykers, S.; Allen, R.; Galinsky, I.; et al. A first-in-human phase I trial of NKG2D chimeric antigen receptor-T cells in AML/MDS and multiple myeloma. J. Clin. Oncol. 2016, 34 (Suppl. 15), TPS3102.
- Curio, S.; Jonsson, G.; Marinović, S. A summary of current NKG2D-based CAR clinical trials. Immunother. Adv. 2021, 1, ltab018.
- Lynn, R.C.; Powell, D.J., Jr. Strain-dependent Lethal Toxicity in NKG2D Ligand-targeted CAR T-cell Therapy. Mol. Ther. 2015, 23, 1559–1561.