Innate immune cells are the early responders to infection and tissue damage. They play a critical role in the initiation and resolution of inflammation in response to insult as well as tissue repair. Following ischemic or non-ischemic cardiac injury, a strong inflammatory response plays a critical role in the removal of cell debris and tissue remodeling. However, persistent inflammation could be detrimental to the heart. Studies suggest that cardiac inflammation and tissue repair needs to be tightly regulated such that the timely resolution of the inflammation may prevent adverse cardiac damage. This involves the recognition of damage; activation and release of soluble mediators such as cytokines, chemokines, and proteases; and immune cells such as monocytes, macrophages, and neutrophils. This is important in the context of doxorubicin-induced cardiotoxicity as well. Doxorubicin (Dox) is an effective chemotherapy against multiple cancers but at the cost of cardiotoxicity. The innate immune system has emerged as a contributor to exacerbate the disease.
- doxorubicin
- doxorubicin-induced cardiotoxicity
- neutrophils
- neutrophil elastase
- innate immunity
- cardiovascular disease
- anthracyclines
1. Introduction
2. Inflammation in the Heart
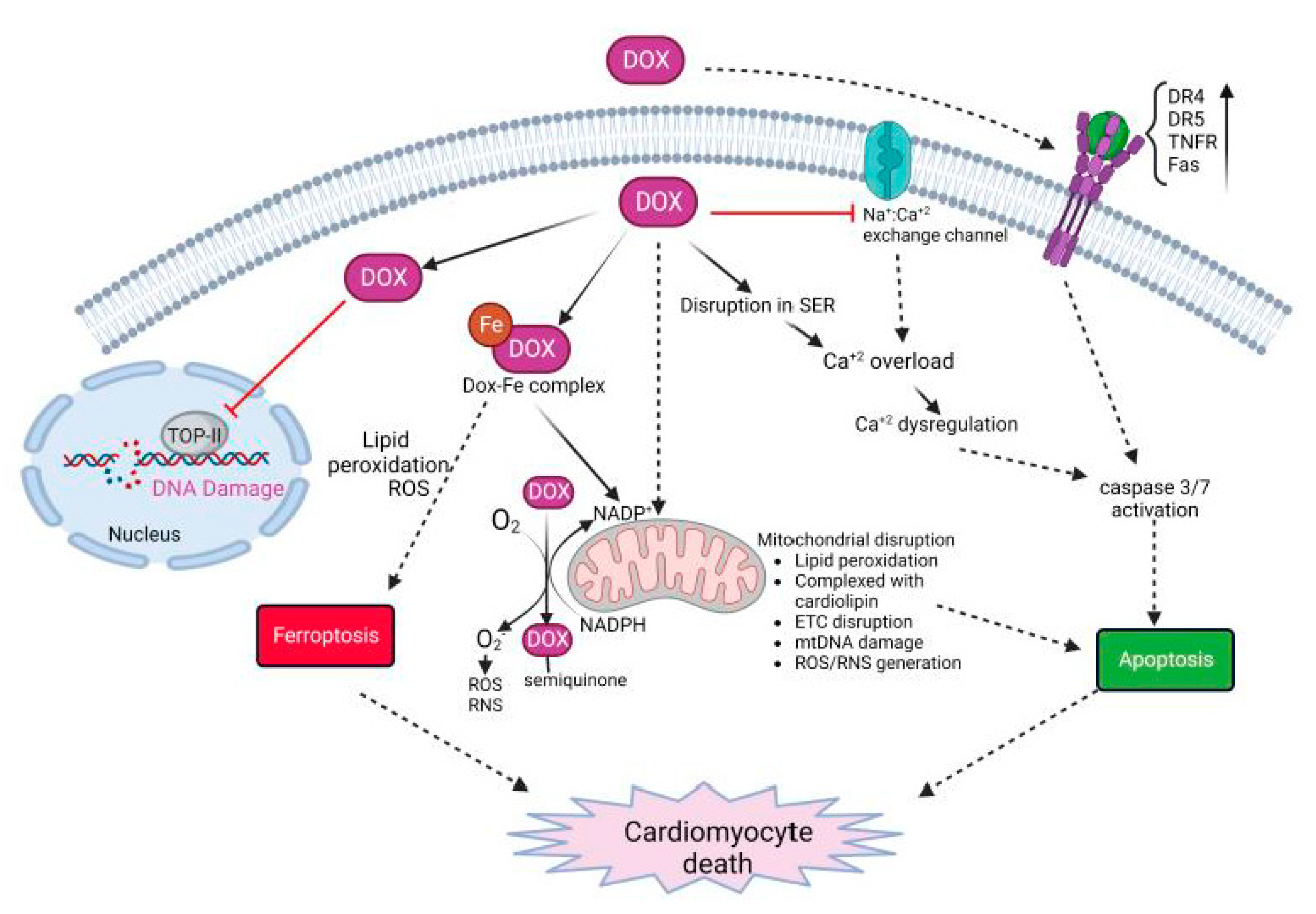
3. Dox-Induced Cardiomyocyte Injury
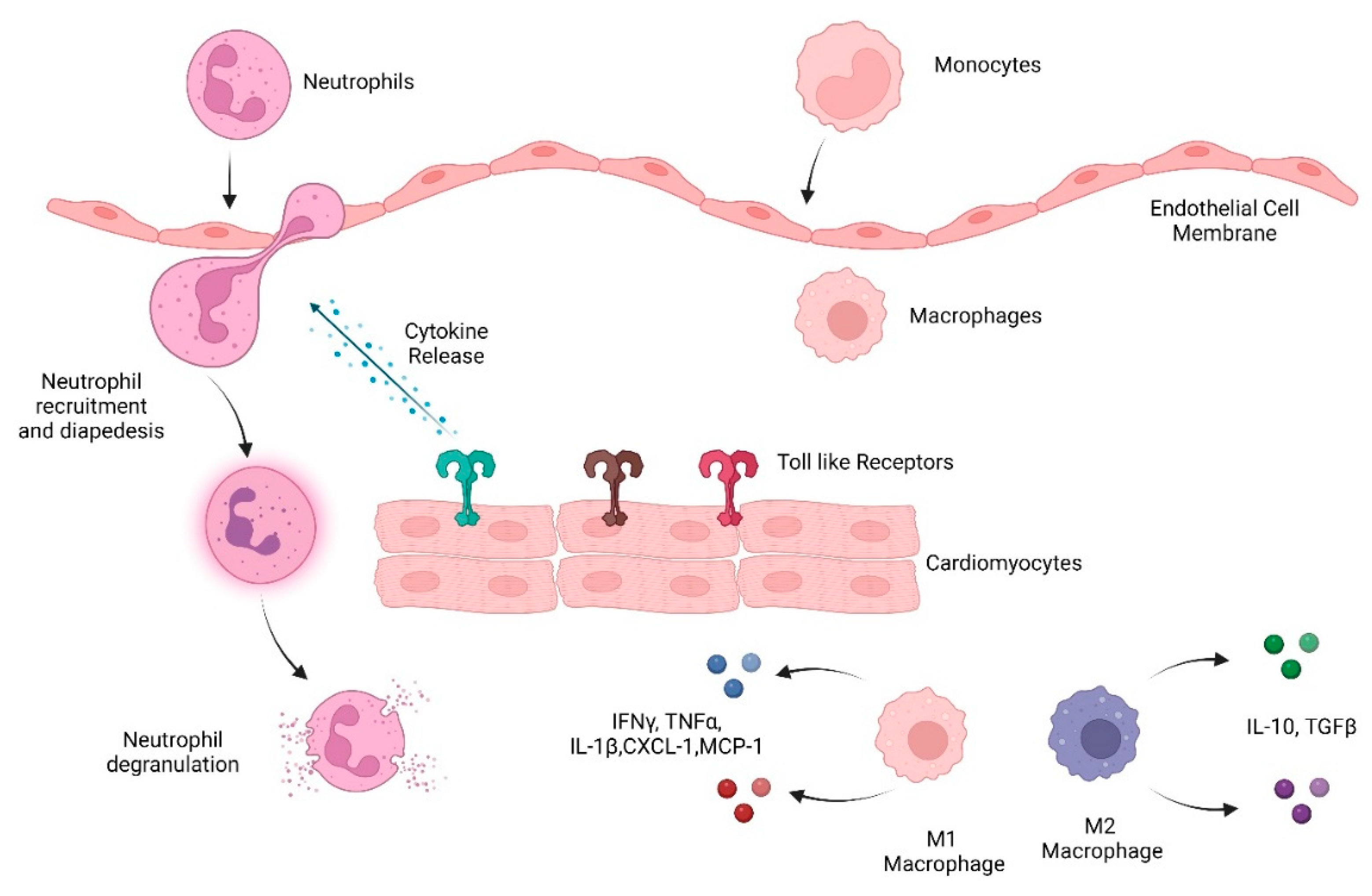
3.1. Role of Immune Response in Dox-Induced Cardiotoxicity
3.1.1. Cytokines/Chemokines
3.1.2. TLRs
3.1.3. Innate Immune Cells
-
Neutrophils
-
Macrophages
-
Invariant natural killer T cells
This entry is adapted from the peer-reviewed paper 10.3390/ijms232314649
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021.
- WHO. Cardiovascular Disease (CVDs). 2021. Available online: https://www.who.int/en/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 23 October 2022).
- Goldsborough, E.; Osuji, N.; Blaha, M.J. Assessment of Cardiovascular Disease Risk: A 2022 Update. Endocrinol. Metab. Clin. 2022, 51, 483–509.
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics—2022 Update: A Report From the American Heart Association. Circulation 2022, 145, e153–e639.
- Noels, H.; Weber, C.; Koenen, R.R. Chemokines as Therapeutic Targets in Cardiovascular Disease. Arter. Thromb. Vasc. Biol. 2019, 39, 583–592.
- Tocchetti, C.G.; Ameri, P.; De Boer, R.A.; D’Alessandra, Y.; Russo, M.; Sorriento, D.; Ciccarelli, M.; Kiss, B.; Bertrand, L.; Dawson, D.; et al. Cardiac dysfunction in cancer patients: Beyond direct cardiomyocyte damage of anticancer drugs: Novel cardio-oncology insights from the joint 2019 meeting of the ESC Working Groups of Myocardial Function and Cellular Biology of the Heart. Cardiovasc. Res. 2020, 116, 1820–1834.
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131.
- Bhatia, S. Genetics of Anthracycline Cardiomyopathy in Cancer Survivors. JACC CardioOncology 2020, 2, 539–552.
- De Angelis, A.; Urbanek, K.; Cappetta, D.; Piegari, E.; Ciuffreda, L.P.; Rivellino, A.; Russo, R.; Esposito, G.; Rossi, F.; Berrino, L. Doxorubicin cardiotoxicity and target cells: A broader perspective. Cardio-Oncology 2016, 2, 2.
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837.
- van Nieuwenhoven, F.A.; Turner, N.A. The role of cardiac fibroblasts in the transition from inflammation to fibrosis following myocardial infarction. Vasc. Pharmacol. 2013, 58, 182–188.
- Baci, D.; Bosi, A.; Parisi, L.; Buono, G.; Mortara, L.; Ambrosio, G.; Bruno, A. Innate Immunity Effector Cells as Inflammatory Drivers of Cardiac Fibrosis. Int. J. Mol. Sci. 2020, 21, 7165.
- Olson, R.D.; Gambliel, H.A.; Vestal, R.E.; Shadle, S.E.; Charlier, H.A.; Cusack, B.J. Doxorubicin Cardiac Dysfunction: Effects on Calcium Regulatory Proteins, Sarcoplasmic Reticulum, and Triiodothyronine. Cardiovasc. Toxicol. 2005, 5, 269–284.
- Wenningmann, N.; Knapp, M.; Ande, A.; Vaidya, T.R.; Ait-Oudhia, S. Insights into Doxorubicin-induced Cardiotoxicity: Molecular Mechanisms, Preventive Strategies, and Early Monitoring. Mol. Pharmacol. 2019, 96, 219–232.
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. Biomed. Pharmacother. 2021, 139, 111708.
- Christidi, E.; Brunham, L.R. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2021, 12, 339.
- Wallace, K.B.; Sardão, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941.
- Mitry, M.A.; Edwards, J.G. Doxorubicin induced heart failure: Phenotype and molecular mechanisms. IJC Heart Vasc. 2015, 10, 17–24.
- Ni, C.; Ma, P.; Wang, R.; Lou, X.; Liu, X.; Qin, Y.; Xue, R.; Blasig, I.; Erben, U.; Qin, Z. Doxorubicin-induced cardiotoxicity involves IFNγ-mediated metabolic reprogramming in cardiomyocytes. J. Pathol. 2019, 247, 320–332.
- Ma, P.; Qin, Y.; Cao, H.; Erben, U.; Ni, C.; Qin, Z. Temporary blockade of interferon-γ ameliorates doxorubicin-induced cardiotoxicity without influencing the anti-tumor effect. Biomed. Pharmacother. 2020, 130, 110587.
- Yu, L.-R.; Cao, Z.; Makhoul, I.; Daniels, J.R.; Klimberg, S.; Wei, J.Y.; Bai, J.P.; Li, J.; Lathrop, J.T.; Beger, R.D.; et al. Immune response proteins as predictive biomarkers of doxorubicin-induced cardiotoxicity in breast cancer patients. Exp. Biol. Med. 2017, 243, 248–255.
- Rassaf, T.; Weber, C.; Bernhagen, J. Macrophage migration inhibitory factor in myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 2014, 102, 321–328.
- Xu, X.; Pang, J.; Chen, Y.; Bucala, R.; Zhang, Y.; Ren, J. Macrophage Migration Inhibitory Factor (MIF) Deficiency Exacerbates Aging-Induced Cardiac Remodeling and Dysfunction Despite Improved Inflammation: Role of Autophagy Regulation. Sci. Rep. 2016, 6, 22488.
- Dufour, J.H.; Dziejman, M.; Liu, M.T.; Leung, J.H.; Lane, T.E.; Luster, A.D. IFN-γ-Inducible Protein 10 (IP-10; CXCL10)-Deficient Mice Reveal a Role for IP-10 in Effector T Cell Generation and Trafficking. J. Immunol. 2002, 168, 3195–3204.
- Clayton, Z.S.; Brunt, V.E.; Hutton, D.A.; Casso, A.G.; Ziemba, B.P.; Melov, S.; Campisi, J.; Seals, D.R. Tumor Necrosis Factor Alpha-Mediated Inflammation and Remodeling of the Extracellular Matrix Underlies Aortic Stiffening Induced by the Common Chemotherapeutic Agent Doxorubicin. Hypertension 2021, 77, 1581–1590.
- Alves, M.T.; Simões, R.; Pestana, R.M.C.; de Oliveira, A.N.; Oliveira, H.H.M.; Soares, C.E.; Sabino, A.D.P.; Silva, L.M.; Gomes, K.B. Interleukin-10 Levels are Associated with Doxorubicin-Related Cardiotoxicity in Breast Cancer Patients in a One-Year Follow-Up Study. Immunol. Investig. 2021, 51, 883–898.
- Ma, Z.-G.; Kong, C.-Y.; Wu, H.-M.; Song, P.; Zhang, X.; Yuan, Y.-P.; Deng, W.; Tang, Q.-Z. Toll-like receptor 5 deficiency diminishes doxorubicin-induced acute cardiotoxicity in mice. Theranostics 2020, 10, 11013–11025.
- Wang, X.; Liu, J.Z.; Hu, J.X.; Wu, H.; Li, Y.L.; Chen, H.L.; Bai, H.; Hai, C.X. ROS-activated p38 MAPK/ERK-Akt cascade plays a central role in palmitic acid-stimulated hepatocyte proliferation. Free Radic. Biol. Med. 2011, 51, 539–551.
- Guo, Z.; Tang, N.; Liu, F.; Yang, Z.; Ma, S.; An, P.; Wu, H.; Fan, D.; Tang, Q. TLR9 deficiency alleviates doxorubicin-induced cardiotoxicity via the regulation of autophagy. J. Cell. Mol. Med. 2020, 24, 10913–10923.
- Nozaki, N.; Shishido, T.; Takeishi, Y.; Kubota, I. Modulation of Doxorubicin-Induced Cardiac Dysfunction in Toll-Like Receptor-2–Knockout Mice. Circulation 2004, 110, 2869–2874.
- Liang, S.; Xinyong, C.; Hongmin, Z.; Jing, W.; Lang, H.; Ping, Z. TLR2 and TLR3 expression as a biomarker for the risk of doxorubicin-induced heart failure. Toxicol. Lett. 2018, 295, 205–211.
- Ye, S.; Su, L.; Shan, P.; Ye, B.; Wu, S.; Liang, G.; Huang, W. LCZ696 Attenuated Doxorubicin-Induced Chronic Cardiomyopathy Through the TLR2-MyD88 Complex Formation. Front. Cell Dev. Biol. 2021, 9, 654051.
- Riad, A.; Bien, S.; Gratz, M.; Escher, F.; Heimesaat, M.M.; Bereswill, S.; Krieg, T.; Felix, S.B.; Schultheiss, H.P.; Kroemer, H.K.; et al. Toll-like receptor-4 deficiency attenuates doxorubicin-induced cardiomyopathy in mice. Eur. J. Heart Fail. 2008, 10, 233–243.
- Wang, L.; Chen, Q.; Qi, H.; Wang, C.; Wang, C.; Zhang, J.; Dong, L. Doxorubicin-Induced Systemic Inflammation Is Driven by Upregulation of Toll-Like Receptor TLR4 and Endotoxin Leakage. Cancer Res. 2016, 76, 6631–6642.
- Sano, S.; Wang, Y.; Ogawa, H.; Horitani, K.; Sano, M.; Polizio, A.H.; Kour, A.; Yura, Y.; Doviak, H.; Walsh, K. TP53-mediated therapy-related clonal hematopoiesis contributes to doxorubicin-induced cardiomyopathy by augmenting a neutrophil-mediated cytotoxic response. JCI Insight 2021, 6.
- Cheng, K.-H.; Contreras, G.P.; Yeh, T.-Y. Potential Role of Neutrophil Extracellular Traps in Cardio-Oncology. Int. J. Mol. Sci. 2022, 23, 3573.
- Bhagat, A.; Shrestha, P.; Jeyabal, P.; Peng, Z.; Watowich, S.S.; Kleinerman, E.S. Doxorubicin-induced cardiotoxicity is mediated by neutrophils through release of neutrophil elastase. Front. Oncol. 2022, 12.
- Kaczmarek, A.; Krysko, O.; Heyndrickx, L.; Aaes, T.L.; Delvaeye, T.; Bachert, C.; Leybaert, L.; Vandenabeele, P.; Krysko, D.V. TNF/TNF-R1 pathway is involved in doxorubicin-induced acute sterile inflammation. Cell Death Dis. 2013, 4, e961.
- Krysko, D.; Kaczmarek, A.; Krysko, O.; Heyndrickx, L.; Woznicki, J.; Bogaert, P.; Cauwels, A.; Takahashi, N.; Magez, S.; Bachert, C.; et al. TLR-2 and TLR-9 are sensors of apoptosis in a mouse model of doxorubicin-induced acute inflammation. Cell Death Differ. 2011, 18, 1316–1325.
- Yang, Y.; Lv, J.; Jiang, S.; Ma, Z.; Wang, D.; Hu, W.; Deng, C.; Fan, C.; Di, S.; Sun, Y.; et al. The emerging role of Toll-like receptor 4 in myocardial inflammation. Cell Death Dis. 2016, 7, e2234.
- Katare, P.B.; Nizami, H.L.; Paramesha, B.; Dinda, A.K.; Banerjee, S.K. Activation of toll like receptor 4 (TLR4) promotes cardiomyocyte apoptosis through SIRT2 dependent p53 deacetylation. Sci. Rep. 2020, 10, 19232.
- Ma, Y.; Zhang, X.; Bao, H.; Mi, S.; Cai, W.; Yan, H.; Wang, Q.; Wang, Z.; Yan, J.; Fan, G.; et al. Toll-Like Receptor (TLR) 2 and TLR4 Differentially Regulate Doxorubicin Induced Cardiomyopathy in Mice. PLoS ONE 2012, 7, e40763.
- Boyd, J.H.; Mathur, S.; Wang, Y.; Bateman, R.M.; Walley, K.R. Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-κB dependent inflammatory response☆. Cardiovasc. Res. 2006, 72, 384–393.
- Liu, L.; Wang, Y.; Cao, Z.; Wang, M.; Liu, X.; Gao, T.; Hu, Q.; Yuan, W.; Lin, L. Up-regulated TLR 4 in cardiomyocytes exacerbates heart failure after long-term myocardial infarction. J. Cell. Mol. Med. 2015, 19, 2728–2740.
- Vannella, K.M.; Wynn, T.A. Mechanisms of Organ Injury and Repair by Macrophages. Annu. Rev. Physiol. 2017, 79, 593–617.
- Lavine, K.J.; Pinto, A.R.; Epelman, S.; Kopecky, B.J.; Clemente-Casares, X.; Godwin, J.; Rosenthal, N.; Kovacic, J.C. The Macrophage in Cardiac Homeostasis and Disease: JACC Macrophage in CVD Series (Part 4). J. Am. Coll. Cardiol. 2018, 72, 2213–2230.
- Williams, J.W.; Giannarelli, C.; Rahman, A.; Randolph, G.J.; Kovacic, J.C. Macrophage Biology, Classification, and Phenotype in Cardiovascular Disease: JACC Macrophage in CVD Series (Part 1). J. Am. Coll. Cardiol. 2018, 72, 2166–2180.
- Lafuse, W.P.; Wozniak, D.J.; Rajaram, M.V.S. Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair. Cells 2020, 10, 51.
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; AlThagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2018, 20, 29–39.
- Zhang, H.; Xu, A.; Sun, X.; Yang, Y.; Zhang, L.; Bai, H.; Ben, J.; Zhu, X.; Li, X.; Wang, Z.; et al. Self-Maintenance of Cardiac Resident Reparative Macrophages Attenuates Doxorubicin-Induced Cardiomyopathy Through the SR-A1-c-Myc Axis. Circ. Res. 2020, 127, 610–627.
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167.
- Ye, J.; Wang, Y.; Xu, Y.; Wang, Z.; Liu, L.; Wang, M.; Ye, D.; Zhang, J.; Yang, Z.; Lin, Y.; et al. Interleukin-22 deficiency alleviates doxorubicin-induced oxidative stress and cardiac injury via the p38 MAPK/macrophage/Fizz3 axis in mice. Redox Biol. 2020, 36, 101636.
- Ye, J.; Huang, Y.; Que, B.; Chang, C.; Liu, W.; Hu, H.; Liu, L.; Shi, Y.; Wang, Y.; Wang, M.; et al. Interleukin-12p35 Knock Out Aggravates Doxorubicin-Induced Cardiac Injury and Dysfunction by Aggravating the Inflammatory Response, Oxidative Stress, Apoptosis and Autophagy in Mice. eBioMedicine 2018, 35, 29–39.
- Huang, K.; Liu, Y.; Tang, H.; Qiu, M.; Li, C.; Duan, C.; Wang, C.; Yang, J.; Zhou, X. Glabridin Prevents Doxorubicin-Induced Cardiotoxicity Through Gut Microbiota Modulation and Colonic Macrophage Polarization in Mice. Front. Pharmacol. 2019, 10, 107.
- Kobayashi, M.; Usui, F.; Karasawa, T.; Kawashima, A.; Kimura, H.; Mizushina, Y.; Shirasuna, K.; Mizukami, H.; Kasahara, T.; Hasebe, N.; et al. NLRP3 Deficiency Reduces Macrophage Interleukin-10 Production and Enhances the Susceptibility to Doxorubicin-induced Cardiotoxicity. Sci. Rep. 2016, 6, 26489.
- Singla, D.; Johnson, T.; Dargani, Z.T. Exosome Treatment Enhances Anti-Inflammatory M2 Macrophages and Reduces Inflammation-Induced Pyroptosis in Doxorubicin-Induced Cardiomyopathy. Cells 2019, 8, 1224.
- Liu, Y.; Wu, M.; Zhong, C.; Xu, B.; Kang, L. M2-like macrophages transplantation protects against the doxorubicin-induced heart failure via mitochondrial transfer. Biomater. Res. 2022, 26, 14.
- Obata, Y.; Ishimori, N.; Saito, A.; Kinugawa, S.; Yokota, T.; Takada, S.; Nakano, I.; Kakutani, N.; Yamanashi, K.; Anzai, T. Activation of invariant natural killer T cells by alpha-galactosylceramide ameliorates doxorubicin-induced cardiotoxicity in mice. Eur. J. Prev. Cardiol. 2020, 27, 2358–2361.