Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Chemistry, Medicinal
Nicotinamide cofactors represent the most common low-potential redox cofactor used in biocatalysis. Although a promising approach, cofactor immobilization depends on the chemical modification of the cofactor to enable the formation of a covalent bond between the cofactor and its flexible linker. The chemical modification of NAD(P)+/NAD(P)H remains challenging, and the major routes used to achieve it are reviewed herein.
- cofactor recycling
- continuous-flow biocatalysis
- biomimetics
- cofactor modification
- nicotinamide
- pyridine nucleotide
1. Introduction
The synthetic structural modification of nicotinamide cofactors is a key technology that enables immobilization. Investigations on the chemical modification of NAD(P)+/NAD(P)H first came to prominence in the 1970s for the purpose of immobilization onto polymeric materials and macromolecular modifiers, such as beads, gels, (nano) fibers and microcapsules [52,53,54]. This approach was used to attach cofactors to surfaces, enabling their incorporation into membrane reactors and affinity chromatographic systems. This was arguably the predecessor of flow biocatalysis. These early systems were further developed to enable the co-immobilization of both the enzyme and cofactor to promote cofactor retention, colocalization and regeneration [55,56].
The modification of nicotinamide nucleosides (and their reduced counterparts) is challenging because the cofactors have likely evolved to achieve chemical stability so as to avoid undesirable biological outcomes. The redox mechanism of the nicotinamide cofactors involves what is often referred to as an electrochemical-chemical-electrochemical (ECE) mechanism, where the oxidation of NAD(P)H to NAD(P)+ involves two single-electron transfers and one proton transfer [57]. Crystal structure analyses of enzyme-cofactor complexes have revealed that the adenine portion of the cofactor NAD+ is generally solvent-accessible when complexed with an enzyme. Modification via chemical tethering at the adenine ring system, therefore, tends to be most the favorable, leading to the least impact upon the cofactor activity [58]. Derivatization at the nicotinamide moiety has been largely avoided to avoid compromising the redox activity of the cofactor.
The specific structural modifications of nicotinamide cofactors are tailored to their intended applications. Despite redox and structural considerations, derivatization has been reported at almost all the available reactive sites on the molecule: the N6, N(1), C8 (adenine), C7 (adenine), 2′/3′ hydroxy (adenine moiety), 2″/3″ hydroxy (nicotinamide moiety) and the pyrophosphate groups (Figure 1). For immobilization, the modification of NAD(P)+ is most often directed towards the primary amine located at the adenine C6 position (often referred to as the N6 position).
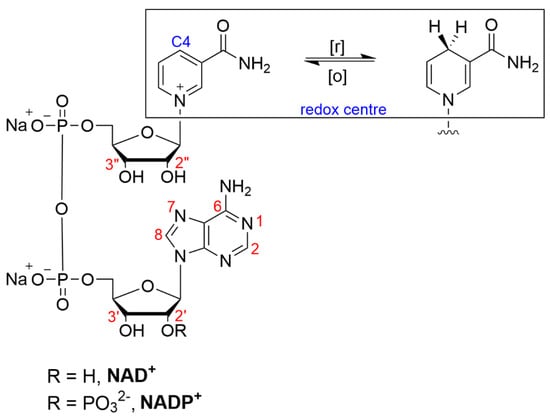
Figure 1. Structure of β-nicotinamide adenine dinucleotide (phosphate), NAD(P)+, indicating sites of modification (red) and redox chemistry (blue).
Structure/activity relationship studies of the N(1) and N6 NAD+ analogues have shown that the adenine moiety protrudes out of the active enzyme pocket, which is why substitutions are preferred in the case of this moiety [59]. Focusing on adenine derivatization, generally, N(1)-substituted NAD+ derivatives are seldom used as potential candidates in biocatalytic processes, as they have been found to substantially reduce the enzymatic activity compared with the unmodified cofactors, in addition to being less stable molecules than their N6-substituted counterparts [59,60,61]. However, generalizations about which positions can be functionalized with the greatest retention of the activity are not always appropriate. For example, it was found that the N(1)-2-hydroxy-carboxypropyl NADP+ was more active than N6-2-hydroxy-carboxypropyl with glucose-6-phosphate dehydrogenase, glutamate dehydrogenase and aldehyde dehydrogenase [62].
There are several synthetic approaches to modification at the N6 position of NAD(P)+. A common method involves an initial substitution at the N(1) position on the purine ring, followed by a base-catalyzed Dimroth rearrangement and a shift of the substituent between the N(1) endocyclic nitrogen and the exocyclic amine (N6) [63]. A second approach involves the phosphate activation of a potentially substituted mononucleotide, followed by the asymmetric condensation of another mononucleotide via imidazolide, morpholidate, or other intermediates. Substitutions used to enable immobilization at other positions, such as the C2, C8 and 2′-OH positions, are less frequently reported.
2. The Dimroth Rearrangement Route
The Dimroth rearrangement route may be favored because the N(1) position is open to facile alkylation by electrophilic reagents under acidic conditions. Iodoacetic acid was used by Mosbach and co-workers to install a carboxymethyl (or, carbamoyl methyl) group at the N(1) position of NAD+ [52] and NADP+, [64] which was followed by the base-mediated translocation of the substituent to the N6 position [55,65]. The rearrangement step was complicated by the need for the prior reduction of the cofactor due to the instability of the oxidized form under basic conditions. Once the reduction and rearrangement of the cofactor were complete, it was re-oxidized enzymatically.
The Dimroth process was later used with propiolactone to form N6-carboxyethyl-NAD+ as well as an epoxide-containing monomer to produce an N6-substituted 2-hydroxyalkylether-NAD+ derivative for the purpose of immobilization (Figure 2) [66,67,68]. NAD+ has also been modified via the same Dimroth rearrangement route, but with ionic end groups that have the potential to interact with membranes charged with the opposite polarity. Here, the cofactor was derivatized with the strongly anionic sulfonatopropyl group using 1,3-propanesultone as a reagent [61,69]. Similarly, strongly cationic N6-2-hydroxy-3-trimethylammonium propyl-NAD+ was prepared in a low yield but showed a good activity [61].
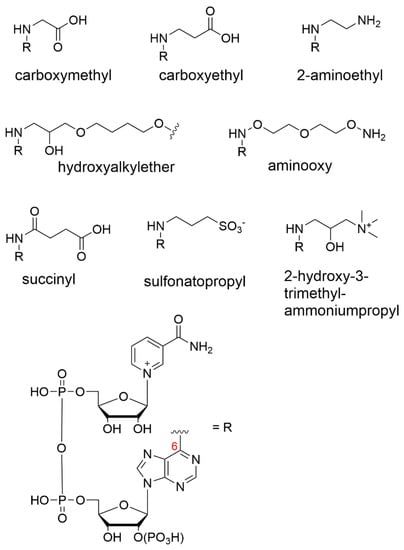
Figure 2. Various N6 nicotinamide cofactor substituents.
The Bückmann group was the first to install an alkylamine at the N6-position using aziridine as the reactive agent, giving rise to 2-aminoethyl substituents (Figure 2). The group suggested that the cofactor reduction/reoxidation step is unnecessary using this route, as the rearrangement reaction from the N(1) to the N6 position can proceed under mildly basic conditions, thus preventing cofactor degradation.
The presence of the 2′-phosphate in NADP+ can complicate its derivatization via the Dimroth rearrangement method. The Okada group reported that the reaction between NADP+ and propiolactone under slightly acidic conditions, followed by alkaline rearrangement, which results in carboxymethylation at the N6 and 2′-phosphate giving a mixture of products [70,71]. This observation however was not reported by Lowe and Mosbach using similar reaction conditions and reagents [64].
3. Phosphate Coupling
Phosphate coupling reactions between two nucleotides are well-known and are often performed between the canonical nucleotides AMP, ADP or ATP and a modified mononucleotide. Typically, anhydrous aprotic solvents are used to prevent the hydrolysis of the activated phosphate back to the native cofactor, and this results in the formation of a symmetrical product containing either purine or pyrimidine bases [72,73]. In terms of the formation of modified pyridine cofactors based on the NAD(P)+ structure, the diphosphate product is not symmetrical, and its synthesis is consequently more nuanced [74,75,76].
The phosphate coupling route permits the modification of a mononucleotide before coupling, such that the derivatization of the adenine can be undertaken on mononucleotides/nucleosides that are less sensitive to base and other harsh reaction conditions. An example of direct derivatization at the N6 position of adenine was reported, [77] where the modified adenine was subsequently coupled to a β-nicotinamide mononucleotide (β-NMN) by a carbodiimide-mediated route to give rise to N6-aminooxy-AMP (Figure 2). The engineered cofactor was later immobilized onto streptavidin-decorated agarose beads. However, the dehydrogenase activity was not measured, as the research was focused on sirtuin-catalyzed deacetylation mechanisms rather than the biocatalytic function.
Various examples of phosphoimidazole activation/coupling routes for the production of NAD(P)+ analogues with alkyl azide/alkyne installation at the N6 position [76] and the purine C2 position have been reported [74,76,78,79]. One approach to this method utilizes 1,1′-carbonyldiimidazole (CDI) to activate β-nicotinamide mononucleotide (β-NMN), forming a phosphoimidazole intermediate. The activated phosphate is then coupled with a 5′-monophosphate—either native adenosine monophosphate (AMP) or an AMP analogue—to achieve the dinucleotide product [80,81]. Morpholidate intermediates can also be used to promote phosphate coupling [72,82,83,84,85].
This entry is adapted from the peer-reviewed paper 10.3390/catal12111454
This entry is offline, you can click here to edit this entry!