Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Tumor-related death is primarily caused by metastasis; consequently, understanding, preventing, and treating metastasis is essential to improving clinical outcomes. Metastasis is mainly governed by the dissemination of tumor cells in the systemic circulation: so-called circulating tumor cells (CTCs). CTCs typically arise from epithelial tumor cells that undergo epithelial-to-mesenchymal transition (EMT), resulting in the loss of cell–cell adhesions and polarity, and the reorganization of the cytoskeleton.
- epithelial-to-mesenchymal transition (EMT)
- circulating tumors cells
- metastasis
1. Introduction
Metastasis, i.e., the spread of cancer cells from their site of origin to distant tissues or organs, is the leading cause of death in almost all solid cancers [1]. However, the cellular and molecular mechanisms that promote tumor cell metastasis are not fully understood. The formation of metastases is mainly driven by the dissemination of cancer cells that are shed from the primary tumor intravasate and circulate in the bloodstream—so-called circulating tumor cells (CTCs). Consequently, CTCs have to resist blood pressure and defend against patrolling immune cells. Thus, extravasation tumor cells have to adapt to the cellular environment of the tissue or organ of the colonization site. Transformation into CTCs requires a number of biochemical and cellular changes that facilitate migration, invasion, resistance to apoptosis, and the modulation of extracellular matrix (ECM) production [2]. During epithelial-to-mesenchymal transition (EMT), ECM is reorganized in such a way that epithelial cells lose cell–cell as well as cell–matrix contacts. Cell adhesion molecules, particularly E-cadherin, which significantly contributes to the epithelial cell junction and apico-basal polarization [3,4,5], are downregulated. Conversely, vimentin, fibronectin, and N-cadherin are upregulated, fostering the transition into a motility state. Overall, cells lose epithelial characteristics while progressively adopting a quasi-mesenchymal phenotype. This cellular program typically is fully reversible and mesenchymal-to-epithelial transition (MET) allows CTCs to regain their initial epithelial phenotype that is required for metastasis formation [5]. In conclusion, EMT and MET—originally implicated in embryogenesis and adult wound healing—play crucial roles in metastatic dissemination by CTCs [6]. Furthermore, EMT is critical for the acquisition of cancer stem cell (CSC)-like properties including self-renewal, resistance to apoptosis, tumor progression, and metastasis. This may result in the generation of cancer cell progenitors with strong drug resistance, e.g., via the expression of membrane protein transporters such as the ATP-binding cassette [7,8].
2. Role of Circulating Tumors Cells with Cancer Stem Cell Properties in Metastasis
Similar to cells of the primary tumor, CTCs are extremely heterogeneous and contain phenotypically and genetically distinct subpopulations including epithelial cells (E-CTCs), epithelial-to-mesenchymal transition cells (EMT-CTCs), hybrid epithelial/mesenchymal cells (EM-CTCs), mesenchymal cells (M-CTCs) and CTCs with stem-cell like properties, termed circulating tumor stem-like cells (CTSCs) [17]. The majority of CTCs, however, cannot survive in the bloodstream and are unable to seed metastases, suggesting that metastatic colonization is a remarkably inefficient process [18]. CTSCs are irregular cells whose phenotype may vary between cancer subtypes and even within the same type of cancer [19]. Owing to their CSC-like properties, CTSCs can escape anoikis [20], are more likely to survive in the bloodstream, and spread metastatically. As they also tend to be more resistant to conventional therapeutic strategies, they are associated with cancer relapse [21]. It is now widely accepted that CTSCs are involved in tumor development, metastasis, and the therapy resistance of solid cancers, including GC, BC, CRC, hepatocellular carcinoma (HCC) and PC. Furthermore, CTSCs and CTCs have been used to diagnose and predict patient outcomes [22,23,24].
Metastasis requires tumor cells to go through a series of events, namely, invasion, intravasation, surviving bloodstream circulation, extravasation, and colonization, commonly referred to as metastatic cascade (Figure 1).
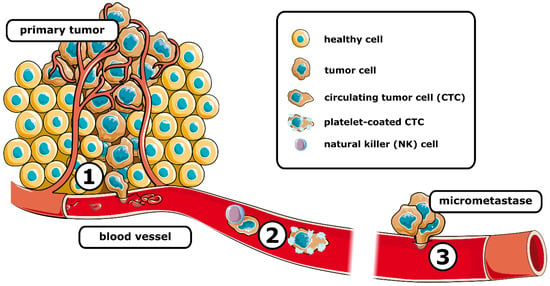
Figure 1. Schematic representation of the metastatic cascade. After intravasation (1) tumor cells travel the systemic circulation (2), where they are susceptible to elimination, e.g., by natural killer cells. Coating by platelets can protect circulating tumor cells. After extravasation, the (3) formation of micrometastases is associated with the colonization of distant tissue. (See text for details).
2.1. Metastatic Cascade
2.1.1. Invasion and Intravasation
The metastatic cascade starts with the invasion of the tumor into the surrounding tissue. Via EMT, epithelial tumor cells can acquire a more invasive, stem-cell-like phenotype allowing them to detach from the primary tumor. The elevated release of MMPs leads to a modulation of ECM and further facilitates invasion [19]. The production of proangiogenic factors such as VEGF promotes angiogenesis. Additionally, CSCs can transdifferentiate into endothelial cells to support the formation of blood vessels [25]. It was shown that endothelial cells in glioblastoma, a highly angiogenic malignancy, shared approximately 60% of their genomic alterations with tumor cells, suggesting that the vascular endothelium originated from the neoplasia. Indeed, it was found that a subcutaneous injection of freshly isolated CD133+/CD31− glioblastoma stem-like cells resulted in human CD31 expression, an endothelial marker, in xenograft tumor tissues of mice [26]. The transdifferentiation of glioblastoma stem-like cells into endothelial-like cells was also confirmed in vitro [27].
Pericytes are multi-functional mural cells of the microcirculation and overexpress the transmembrane receptor endosialin. Thereby, these cells promote cell–cell contact with tumor cells and foster their migration through the endothelium. In the TME, pericytes are also the main source of IL33, and thus responsible for recruiting tumor-associated macrophages (TAMs) via the IL33-ST2 signaling pathways [28,29]. Moreover, monocytes recruited by CCR2 signaling can differentiate into TAMs. CXCL12 released by perivascular fibroblasts facilitates the co-migration of tumor cells and TAMs into blood vessels. The differentiation of TAMs into perivascular macrophages stimulates vascular leakage, further promoting intravasation [30].
2.1.2. Surviving Bloodstream Circulation
After intravasation, CTCs face a hostile environment that poses both physical and biological threats [31]. Above all, circulating in the bloodstream subjects cells to shear stress. As CTCs are poorly adapted to this kind of mechanical force [32], most of them will be directly destroyed or enter apoptosis. Additionally, CTCs have to withstand the constant attacks of immune cells. The interplay of internal and external factors critically determines whether a cell can survive in the bloodstream. Internal factors include genetic alterations and the abnormal gene expression of, e.g., immunomodulatory factors and apoptosis inhibitory factors such as survivin or stem cell-like characteristics. External factors involve immune cells, cytokines, platelets, and circulating tumor microemboli that may protect CTCs from cellular stress. Among others, tissue hypoxia and autophagy may also impact cell survival [33]. CTCs can undermine immunological clearance through several different strategies. They often overexpress immunosuppressive proteins such as the immune checkpoint PD-L1, e.g., in BC, esophageal cancer, CRC, and PC [34,35]. The immune checkpoint V-domain Ig suppressor of T-cell activation (VISTA) is upregulated in lymphocytes from metastatic melanoma patients, suggesting that VISTA might be implicated in metastasis [36]. In addition, VISTA was highly expressed in advanced stages of ovarian cancer and particularly dominant in lymph node metastases. Recently, it has been shown that VISTA inhibits T-cell effector functions and the release of IL2 from CD4+ T cells by suppressing granzyme B release from cytotoxic T cells [37,38]. Moreover, platelets seem to have a significant role in the immune evasion of CTCs, as these cells attract platelets by the expression of coagulation factors, for instance, thromboplastin. Being fully covered with platelets protects CTCs from NK and other immune cells and reduces the shear stress on CTCs [39,40]. Platelet-derived TGF-β and direct platelet–tumor cell contact activates the TGF-β/Smad and NF-κB pathways in a synergistic manner, which promotes the acquisition of a mesenchymal-like phenotype [41]. Furthermore, CD47 expressed on the surface of CTCs inhibits the interaction with macrophage-expressed signal-regulating protein alpha (SIRPα) and subverts the phagocytic machinery [42,43]
2.1.3. Extravasation and Colonization
The small number of CTCs that are able to withstand blood circulation can eventually settle in a distant organ after extravasation [44]. In 1889, Paget shaped the seed and soil hypothesis, according to which CTCs and the host organ must have a certain degree of compatibility to allow for the formation of metastases [45]. Albeit challenged over the last century, Paget’s theory seems to be supported by recent research on the mechanisms of metastatic diseases. However, the initial theory has been extended by the concept of pre-metastatic niche formation: prior to the dissemination of tumor cells, the primary tumor can induce the formation of a supportive and receptive tissue microenvironment via the production of tumor-derived secreted factors VEGF-A, tumor necrosis factor α (TNF-α) and TGF-β. As a result of this preparation, tumor cells settle more effectively owing to an accumulation of compromised immune cells and ECM [46]. Some solid cancers have preferred organs for metastasis, e.g., PC prefers to metastasize to the liver and peritoneum, with more than 40% preference for those organs and about 13% preference for the lung. CRC and GC are even more inclined to metastasize to the liver with about 70% and 48%, respectively, making the liver the most common organ for those solid cancers to metastasize [47,48,49]. Prior to extravasation, CTCs adhere to the endothelium via CD44, integrin β1, integrin αvβ3, and α5β1 [19,50]. CTCs release glycocalyx that enhances integrin binding in order to promote intravascular adhesion and extravasation [51]. It has been demonstrated that metastatic colonies originate from intravascular tumor cells [52], which in the last step of the metastatic cascade, the extravasation, migrate through the endothelium of the blood vessel and colonize the pre-metastatic niche [44]. In addition, exosomes, which facilitate the communication of primary tumor cells with the microenvironment of distant organs, are implicated in pre-metastatic niche formation [53]. A role of exosomes as a key factor for the (epigenetic) reprogramming of cells in targeted organs has been reviewed elsewhere [54]. It seems likely that CTCs need to acquire stem cell-like properties for successful colonization. Recently, cellular and transcriptomic changes were observed during in vivo metastasis formation in a clonal outgrowth model of patient-derived CRC organoids in mice. Peculiarly, micrometastatic lesions were devoid of CSCs, but on the other hand, de novo CSCs were indeed present in overt metastases [55]. After extravasation, metastatic colonization is further stimulated by MET, which triggers the re-expression of E-cadherin and the downregulation of Prrx1, which subsequently decreases Snail 1 expression and leads to the re-acquisition of an epithelial phenotype and epithelial morphology [56,57].
This entry is adapted from the peer-reviewed paper 10.3390/cancers14225483
This entry is offline, you can click here to edit this entry!