Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Emergency Medicine
Novel oral anticoagulants (NOACs) are drugs selective for thrombin or activated factor Xa approved for the prevention and treatment of many thromboembolic conditions. They included venous thromboembolism in patients undergoing orthopedic surgery, such as hip or knee arthroplasty, atrial fibrillation, stroke prevention in nonvalvular patients, and pulmonary embolism. NOACs, such as apixaban, edoxaban, dabigatran, rivaroxaban and betrixaban, have become an alternative to vitamin K anticoagulants (i.e., warfarin).
- novel oral anticoagulants
- NOACs
- gastrointestinal bleeding
- edoxaban
- apixaban
1. Pathophysiology of Gastrointestinal Bleeding under NOACs Treatment
The digestive tract is endowed with a rich intra and submucosal vascularization. However, even in conditions of wellbeing, the gastrointestinal mucosa is regularly damaged. In this regard, endoscopic data on healthy volunteers allowed us to note how gastric and small bowel erosions are present in 5–10% and 10–15% of cases, respectively. This mucosal vulnerability, which is the expression of the impact of acid and digestive enzymes such as amylase, trypsin and pepsin, as well as exogenous bacterial flora, makes intestinal vascularization prone to clinical or sub-clinical bleeding. This bleeding tendency is, however, modified by the intake and the consequent effect of anticoagulants, and various mechanisms have been described (Figure 1): (a) topical anticoagulant effect, (b) systemic anticoagulant effect, (c) caustic action at local level, (d) topical biological effect not correlated to coagulation (inhibition of mucosal healing), or (e) a combination of these [29,30,31]. While, for warfarin, which is 95% absorbed, only a systemic effect has been described, and the unabsorbed fraction does not appear to cause topical damage. For NOACs, more mucosal damage mechanisms have been highlighted. First of all, the absorption of NOACs is variable. For example, the bioavailability of rivaroxaban is 60–80%, and that of apixaban is 50% higher than that of dabigatran. The latter, taken as a prodrug, has an oral bioavailability of just 6%, while the remainder passes through the intestine and, before being excreted in the feces, is two-thirds converted into an active drug by intestinal esterases. Despite the different levels of bioavailability, an important amount of active drug is excreted with the feces and, therefore, could have a local, as well as systemic, effect on a pre-existing mucosal vulnerability [11,31,32,33]. Another factor that must be taken into account when considering the pathophysiology of gastrointestinal bleeding in these anticoagulated patients is the knowledge that NOACs are substrates of a key efflux pump present along the gut, namely, the permeability glycoprotein (P-gp) [34,35]. This transporter, also called the ATP-binding cassette (ABC) subfamily member 1 (ABCB1) or multidrug resistance protein 1 (MDR1), is likely the actor of a protective mechanism against xenobiotics. It is present in different organs (e.g., kidney, liver, and blood-brain barrier) and, particularly, it is widely expressed along the intestinal epithelium, where it pumps such substances (e.g., drugs and toxins) back into the lumen [36,37]. Indeed, by transporting a series of substrates across intra- and extra-cellular membranes, P-gp is often involved in drug–drug interactions, as some drugs can either act as a substrate, an inducer, or an inhibitor of this glycoprotein. If NOACs are administered together with a P-gp inhibitor, the decrease in luminal efflux of the drug by this transporter will translate into a higher blood concentration of anticoagulant, thus increasing the bleeding risk [38]. Furthermore, P-gp is expressed in different concentrations along the bowel, with maximum expression in the distal ileum and minimum in the duodenal level. Consequently, the more distal segments appear to have a greater efflux of anticoagulant at the intraluminal level [39]. In this regard, some studies on surgical patients have been conducted on the effect of NOACs in patients undergoing bypass or major gastrointestinal resections, showing how these surgeries on the proximal gastrointestinal tract led to greater exposure to the anticoagulant in the more distal segments, which would then result in more sensitivity to NOAC topical damage [40]. Moreover, different in vitro studies [34,41] have been conducted so as to better characterize the expression of P-gp at the level of the intestinal epithelium.
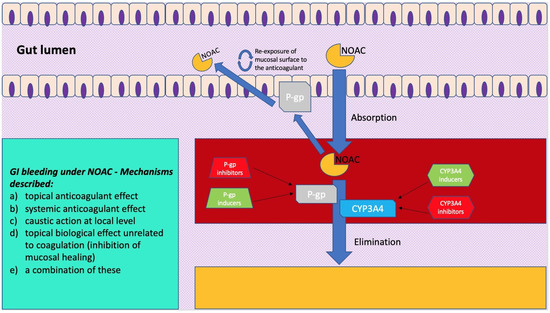
Figure 1. A simplification model of how molecular mechanisms related to pharmacokinetics of novel oral anticoagulants (NOACs) exert a clinical impact on gastrointestinal bleeding. On the left is a scheme showing mechanisms described for gastrointestinal bleeding under NOAC treatment.
2. The Role on Gastrointestinal Bleeding of ABC Transporters and Permeability Glycoprotein (P-gp)
Englund et al. [41] quantified the expression of a series of transporters of the ABC group along the gut using biopsies from volunteers undergoing routine examination and compared the results with in vitro analysis on an intestinal model derived from human colorectal adenocarcinoma cells. In this Swedish study [41], the authors found that P-gp levels were up to five-fold higher in the ileum compared to the duodenum or colon. Particularly, this intestinal model, which has been used in more than one study on NOAC gut-related pharmacokinetics, is composed of a particular cell line (Caco-2) [42,43] cultured under specific conditions so as to differentiate and (although being derived from the colon) resemble in phenotype the enterocytes of the small intestine to act as intestinal model, and expresses, among a number of transporters and enzymes, P-gp and breast cancer resistance protein (BCRP), which are both involved in NOAC pharmacokinetics. A comparative study by Gnoth et al. [44] on transport characteristics of rivaroxaban, both in vivo, using P-gp double-knockout mice vs. wild-type, and in vitro, studying the anticoagulant bidirectional efflux through Caco-2 model, wild-type and P-gp overexpressing cells, stresses that this NOAC is a substrate, but not an inhibitor, of P-gp, and that this mechanism can lead to drug–drug interactions (DDI). Hodin et al. [34] conducted a similar study using the same Caco-2 intestinal model to investigate in the case of all the NOACs’ transport via P-gp and BCRP. Three different ABC carrier-mediated transport profiles are identified: predominantly P-gp-dependent transport for dabigatran, preferential BCRP-dependent transport for apixaban, and approximately equivalent P-gp and BCRP-mediated transport regarding edoxaban and rivaroxaban. Zhang et al. [45], working on animal models, administered active charcoal to bile duct-cannulated dogs and rats receiving an intravenous dose of radiolabeled apixaban and studied drug transporter knockout rats and highlighted that, not only was apixaban undergoing intestinal excretion, but there was also a mechanism of entero–enteric recirculation of the anticoagulant via a mechanism of luminal excretion through efflux pumps followed by reabsorption. This finding gives further strength to the direct, topical damage of NOACs within the gut that has been described in literature. As mentioned before, a potentiation of systemic and topical damage can occur when NOACs are co-administered with drugs that are known substrates, inhibitors, or inducers of P-gp, BCRP, or cytochrome CYP3A4 (Figure 1).
3. Pharmacokinetic Features of NOACs
NOACs are characterised by both different volumes of distribution and different binding to plasma proteins. The volume of distribution of apixaban is small, suggesting distribution mainly in the systemic circulation, with limited extravascular localisation. In contrast, dabigatran is characterised by high hydrophilicity, poor plasma protein binding, and essential renal clearance, features that make this NOAC the only haemo-dialisable one. In this regard, it should be emphasised that dialysis is effective if performed within the first few hours of administration, otherwise the drug’s volume of distribution (60 L) prevents dialysis clearance of approximately 12 L when distribution is complete. An important pharmacokinetic feature distinguishing NOACs is the route of elimination, which is essentially renal for dabigatran, while it is both hepatic and renal for rivaroxaban, apixaban, and edoxaban. This leads to variations in the dosage and choice of NOACs based on the patient’s pathophysiological and demographic characteristics. For example, dabigatran is contraindicated in patients with a creatinine clearance <30 mL/min. Rivaroxaban and apixaban can be used with caution, always reducing the dose and monitoring the patient’s renal function. In patients with moderate renal impairment (creatinine clearance between 30 and 50 mL/min), dabigatran and rivaroxaban can be used at reduced doses, whereas apixaban, which is less renally eliminated, can be used at normal doses, at least in patients aged ≤80 years and weighing >60 kg.
All NOACs are, however, contraindicated in patients with severe hepatic impairment, while dose reduction is recommended for apixaban, rivaroxaban, and edoxaban when co-administered with potent CYP3A4 inhibitors, unlike dabigatran, which is not metabolised by cytochromes. Among the characteristics of NOACs, the elimination half-life of approximately 12 h to 14 h deserves consideration, a characteristic that suggests a dosage with double daily administration. The ratio between the maximum and minimum steady-state concentration in single daily administration is 4.5 for dabigatran, 10 for rivaroxaban, 10 for apixaban and 10–30 for edoxaban. The higher the ratio, the greater the fluctuation in plasma levels over 24 h.
The clinical consequences of these fluctuations can obviously lead to bleeding at the peak or thromboembolic events at the lowest concentrations. It is therefore plausible to try to minimise these variations by opting for twice a day administration and extended/controlled release forms. This option is also justified by the fact that a bid administration should benefit from a more constant maintenance of the plasma concentration of the NOACs and their actions, even in the event of variable drug exposure due to suboptimal treatment adherence. However, for rivaroxaban, it was preferred to opt for single administration. The high C-max levels with rivaroxaban, together with kinetics that are non-linear at doses >10 mg, which require intake with food, result in significant variability in peak concentrations. In this regard, a cross-over study was conducted comparing the pharmacokinetic and pharmacodynamic profile of apixaban 2.5 mg bid and rivaroxaban 10 mg/day 30 in the same patients. A mean C-max variability of 46% and 23% can be observed with rivaroxaban and apixaban, respectively. This extreme variability of rivaroxaban could be associated with the increased risk of gastrointestinal bleeding, compared to warfarin, observed in the ROCKET AF [27] clinical trial. To summarize, rivaroxaban [52] is mainly absorbed in the proximal small intestine, with dose-dependent bioavailability, and it is not linear with the dose administered. At the 10 mg dose, the estimated bioavailability is 80–100%, compared to 66% at the 20 mg dose, when administered on an empty stomach. The presence of food, probably by increasing its solubilisation and dissolution, significantly increases the bioavailability of rivaroxaban 20 mg, while also reducing the inter-individual variability of its plasma concentrations. It is important to remember that food intake implies a meal of at least 1300 Cal with 30–40% fat content. This is why it is essential to take rivaroxaban after meals. Apixaban [53] is predominantly absorbed in the distal part of the small intestine and in the ascending colon, reaching its peak concentration (C-max) 2–3 h after oral intake. Its bioavailability is approximately 50%, and approximately 35% of the unabsorbed portion is excreted with feces. Unlike rivaroxaban, intestinal absorption of apixaban is not affected by the presence of food. Similar considerations can be made regarding the absorption of edoxaban, which reaches C-max after 2 h and is not affected by the presence of food.
4. Pharmacodynamics of NOACs: Efficacy and Safety Considerations
From a pharmacodynamic point of view, NOACs have a direct effect that is maximal 2–3 h after their administration in accordance with the time to peak concentration and the direct correlation between plasma concentrations and anticoagulant effect. These characteristics distinguish them from warfarin, which, by inhibiting the activation of several coagulation factors, takes 3–5 days to manifest its anticoagulant action. This difference means that the initiation of NOAC treatment does not require a period of pre-treatment with heparin (‘bridging’). The same reasoning applies to the reversibility of the effect, which is much faster for NOACs, both because of their short half-life and the reversibility of their mechanism of action, than for warfarin. A detailed analysis of the bleedings observed in the different districts shows important differences between the different NOACs compared to warfarin. Dabigatran 150 mg bid (but not 110 mg bid) had a similar incidence to warfarin in cases of major bleeding (RE-LY) [25]. In the ROCKET AF [27] study, a comparable incidence of major bleeding was observed between rivaroxaban and warfarin. In contrast, the risk of bleeding was significantly lower for both apixaban (ARISTOTLE) [28] and edoxaban (ENGAGE) [26] compared to warfarin. Importantly, all NOACs demonstrated superiority over warfarin in intracranial haemorrhage. With regard to gastrointestinal bleeding, differences were observed between the various NOACs compared to warfarin. The administration of dabigatran 150 mg bid is associated with a higher and more significant incidence of gastrointestinal bleeding than warfarin. In particular, bleeding in patients treated with dabigatran was mostly referred to the lower part of the small intestine (53%) compared to those observed with warfarin (25%), in agreement with the activation of dabigatran from prophylactic to drug that occurs during the gastrointestinal route. Rivaroxaban causes a significantly higher increase in gastrointestinal bleeding than warfarin, probably attributable to its single-dose dosage with very high peak concentrations and extreme variability. In accordance with its site of absorption, bleeding occurs mainly in the proximal part of the small intestine. In contrast to dabigatran and rivaroxaban, apixaban is not significantly different in the risk of gastrointestinal bleeding compared to warfarin in terms of both incidence and site of bleeding. As for edoxaban, results from the ENGAGE AF-TIMI 48 [26] study show that the risk of gastrointestinal bleeding is significantly higher at high doses (60 mg/day) and lower at low doses (30 mg/day). In accordance with its kinetic profile, no differences are observed compared to warfarin in gastrointestinal bleeding sites. In clinical practice, the results of a recent Food and Drug Administration analysis report confirm an increased risk of gastrointestinal bleeding for rivaroxaban and dabigatran, in agreement with clinical studies. The authors of this pharmacovigilance study conclude that apixaban, among the NOACs, appears to be the drug with the best safety profile in terms of both major bleeding and gastrointestinal bleeding with equal efficacy. It is important to note that the pharmacovigilance data confirm those reported in the registration studies.
This entry is adapted from the peer-reviewed paper 10.3390/ijms232213955
This entry is offline, you can click here to edit this entry!