In bioinformatics, a Gene Disease Database is a systematized collection of data, typically structured to model aspects of reality, in a way to comprehend the underlying mechanisms of complex diseases, by understanding multiple composite interactions between phenotype-genotype relationships and gene-disease mechanisms. Gene Disease Databases integrate human gene-disease associations from various expert curated databases and text mining derived associations including Mendelian, complex and environmental diseases.
- bioinformatics
- model
- text mining
1. Introduction
Experts in different areas of biology and bioinformatics have been trying to comprehend the molecular mechanisms of diseases to design preventive and therapeutic strategies for a long time. For some illnesses, it has become apparent that it is the right amount of animosity is made for not enough to obtain an index of the disease-related genes but to uncover how disruptions of molecular grids in the cell give rise to disease phenotypes.[1] Moreover, even with the unprecedented wealth of information available, obtaining such catalogues is extremely difficult.
Genetic Broadly speaking, genetic diseases are caused by aberrations in genes or chromosomes. Many genetic diseases are developed from before birth. Genetic disorders account for a significant number of the health care problems in our society. Advances in the understanding of this diseases have increased both the life span and quality of life for many of those affected by genetic disorders. Recent developments in bioinformatics and laboratory genetics have made possible the better delineation of certain malformation and mental retardation syndromes, so that their mode of inheritance can be understood. This information enables the genetic counselor to predict the risk for occurrence of a large number of genetic disorders.[2] Most genetic counseling is done, however, only after the birth of at least one affected individual has alerted the family to their predilection for having children with a genetic disorder. The association of a single gene to a disease is rare and a genetic disease may or may not be a transmissible disorder.[3] Some genetic diseases are inherited from the parent’s genes, but others are caused by new mutations or changes to the DNA. In other occurrences, the same disease, for instance, some forms of carcinoma or melanoma, may stem from an inbred condition in some people, from new changes in other people, and from non-genetic causes in still other individuals.[4]
There are more than six thousand known single-gene disorders (monogenic), which occur in about 1 out of every 200 births.[5] As their term suggests, these diseases are caused by a mutation in one gene. By contrast, polygenic disorders are caused by several genes, regularly in combination with environmental factors.[6] Examples of genetic phenotypes include Alzheimer's disease, breast cancer, leukemia, Down syndrome, heart defects, and deafness; therefore, cataloguing to sort out all the diseases related to genes is needed.
1.1. Challenges with Creation
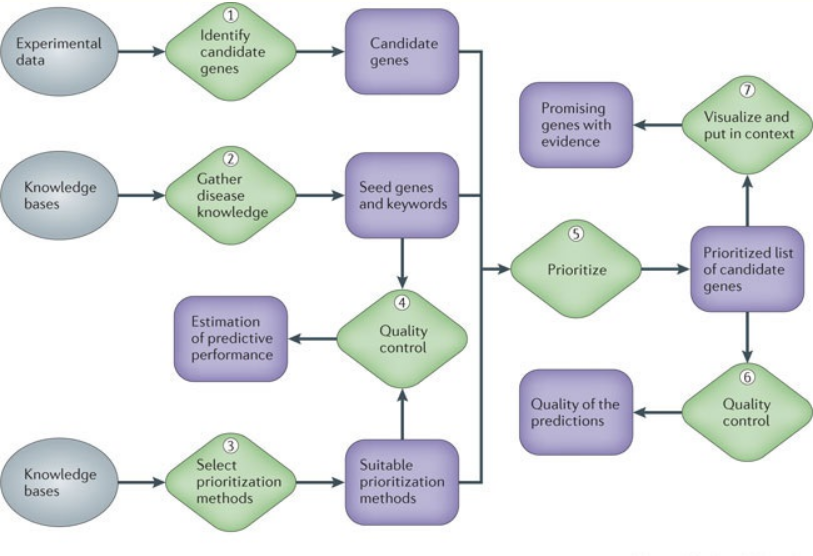
At different stages of any gene disease project, molecular biologists need to choose, even after careful statistical data analysis, which genes or proteins to investigate further experimentally and which to leave out because of limited resources. Computational methods that integrate complex, heterogeneous data sets, such as expression data, sequence information, functional annotation and the biomedical literature, allow prioritizing genes for future study in a more informed way. Such methods can substantially increase the yield of downstream studies and are becoming invaluable to researchers. So one of the main concerns in biological and biomedical research is to recognise the underlying mechanisms behind this intricate genetic phenotypes. Great effort has been spent on finding the genes related to diseases[7]
However, increasingly evidences point out that most human diseases cannot be attributed to a single gene but arise due to complex interactions among multiple genetic variants and environmental risk factors. Several databases have been developed storing associations between genes and diseases such as the Comparative Toxicogenomics Database (CTD), Online Mendelian Inheritance in Man (OMIM), the genetic Association Database (GAD) or the Disease genetic Association Database (DisGeNET). Each of these databases focuses on different aspects of the phenotype-genotype relationship, and due to the nature of the database curation process, they are not complete, but in a way they are fully complementary between each other.[8]
2. Types of Databases
Essentially, there are four types of databases: curated databases, predictive databases, literature databases and integrative databases[5]
2.1. Curated Databases
The term curated data refers to information, that may comprise the most sophisticated computational formats for structured data, scientific updates, and curated knowledge, that has been composed and prepared under the regulation of one or more experts considered to be qualified to engage in such an activity[9] The implication is that the resulting database is of high quality. The contrast is with data which may have been gathered through some automated process or using particularly low or inexpert unsupported data quality and possibly untrustworthy.[9] Some of the most common examples include: CTD and UNIPROT.
The Comparative Toxicogenomics Database (CTD)
The Comparative Toxicogenomics Database, helps to understand about the effects of environmental compounds on human health by integrating data from curated scientific literature to describe biochemical interactions with genes and proteins, and links between diseases and chemicals, and diseases and genes or proteins.[10] CTD contains curated data defining cross-species chemical–gene/protein interactions and chemical– and gene–disease associations to illuminate molecular mechanisms underlying variable susceptibility and environmentally influenced diseases. These data deliver insights into complex chemical–gene and protein interaction networks. One of the main sources in this Database is curated information from OMIM.[10]
CTD is a unique resource where bioinformatics specialists read the scientific literature and manually curate four types of core data:
- Chemical-gene interactions
- Chemical-disease associations
- Gene-disease associations
- Chemical-phenotype associations
The Universal Protein Resource (UNIPROT)
The Universal Protein Resource (UniProt) is an inclusive resource for protein sequence and annotation data. It is a comprehensive, first-class and freely accessible database of protein sequence and functional information, that has many entries being derived from genome sequencing projects. It contains a large amount of information about the biological function of proteins derived from the study literature, which can hint to a direct connection between gene-protein-disease.[11]
2.2. Predictive Databases
A predictive database is one based on statistical inference. One particular approach to such inference is known as predictive inference, but the prediction can be undertaken within any of the several approaches to statistical inference. Indeed, one description of biostatistics is that it provides a means of transferring knowledge about a sample of a genetic population to the whole population (genomics), and to other related genes or genomes, which the same as prediction over time is not necessarily.[13] When information is transferred across time, often to specific points in time, the process is known as forecasting. Three of the main examples of databases that can be considered in this category include: The Mouse genome Database (MGD), The Rat genome Database (RGD), OMIM and the SIFT Tool from Ensembl.[5]
The Mouse genome Database (MGD)
The Mouse genome Database (MGD) is the international community resource for integrated genetic, genomic and biological data about the laboratory mouse. MGD provides full annotation of phenotypes and human disease associations for mouse models (genotypes) using terms from the Mammalian Phenotype Ontology and disease names from OMIM.[14]
The Rat Genome Database (RGD)
The Rat Genome Database (RGD) began as a collaborative effort between leading research institutions involved in rat genetic and genomic research. The rat continues to be extensively used by researchers as a model organism for investigating the biology and pathophysiology of disease. In the past several years, there has been a rapid increase in rat genetic and genomic data.[15] This explosion of information highlighted the need for a centralized database to efficiently and effectively collect, manage, and distribute a rat-centric view of this data to researchers around the world. The Rat Genome Database was created to serve as a repository of rat genetic and genomic data, as well as mapping, strain, and physiological information. It also facilitates investigators research efforts by providing tools to search, mine, and predict this data.[15]
Data at RGD that is useful for researchers investigating disease genes include disease annotations for rat, mouse and human genes. Annotations are manually curated from the literature, or downloaded via automated pipelines from other disease-related databases. Downloaded annotations are mapped to the same disease vocabulary used for manual annotations to provide consistency across the dataset. RGD also maintains disease-related quantitative phenotype data for the rat (PhenoMiner).[16]
The Online Mendelian Inheritance in Man (OMIM)
Supported by the NCBI, The Online Mendelian Inheritance in Man (OMIM) is a database that catalogues all the known diseases with a genetic component, and predicts their relationship to relevant genes in the human genome and provides references for further research and tools for genomic analysis of a catalogued gene.[17] OMIM is a comprehensive, authoritative compendium of human genes and genetic phenotypes that is freely available and updated daily. The database has been used as a resource for predicting relevant information to inherited conditions.[17]
Ensembl SIFT tool
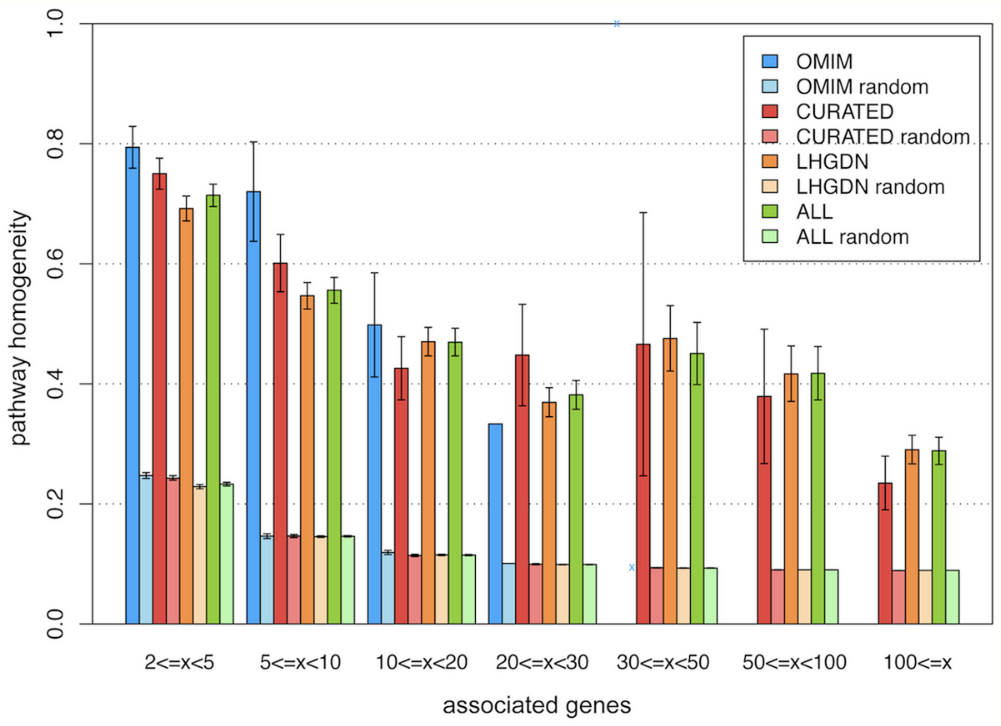
This one of the largest resources available for all genomic and genetic studies, it provides a centralized resource for geneticists, molecular biologists and other researchers studying the genomes of our own species and other vertebrates and model disease organisms. Ensembl is one of several well-known genome browsers for the retrieval of genomic-disease information. Ensembl imports variation data from a variety of different sources, Ensembl predicts the effects of variants.[18] For each variation that is mapped to the reference genome, each Ensembl transcript is identified that overlap the variation. Then it uses a rule-based approach to predict the effects that each allele of the variation may have on the transcript. The set of consequence terms, defined by the Sequence Ontology (SO) can be currently assigned to each combination of an allele and a transcript. Each allele of each variation may have a different effect in different transcripts. A variety of different tools are used to predict human mutations in the Ensembl database, one of the most widely used is SIFT, that predicts whether an amino acid substitution is likely to affect protein function based on sequence homology and the physic-chemical similarity between the alternate amino acids. The data provided for each amino acid substitution is a score and a qualitative prediction (either 'tolerated' or 'deleterious'). The score is the normalized probability that the amino acid change is tolerated so scores near 0 are more likely to be deleterious. The qualitative prediction is derived from this score such that substitutions with a score < 0.05 are called 'deleterious' and all others are called 'tolerated'. SIFT can be applied to naturally occurring nonsynonymous polymorphisms and laboratory-induced missense mutations, that will lead to build relationships in phenotype characteristics, proteomics and genomics.[18]
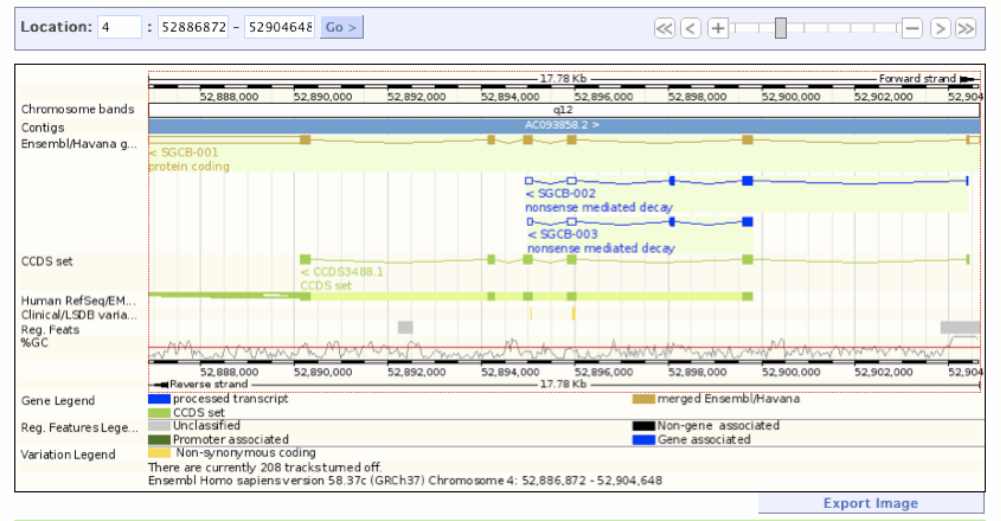
Ensembl release58 sgcb screenshot. https://handwiki.org/wiki/index.php?curid=1311818
2.3. Literature Databases
This sort of databases summarize books, articles, book reviews, dissertations, and annotations about gene-disease databases. Some of the following are examples of this type: GAD, LGHDN and BeFree Data.
Genetic Association Database (GAD)
The Genetic Association Database is an archive of human genetic association studies of complex diseases. GAD is primarily focused on archiving information on common complex human disease rather than rare Mendelian disorders as found in the OMIM. It includes curated summary data extracted from published papers in peer reviewed journals on candidate gene and genome Wide Association Studies (GWAS).[19] The GAD was frozen as of 09/01/2014 but is still available for download.[20]
Literature-derived human gene-disease network (LHGDN)
The literature-derived human gene-disease network (LHGDN) is a text mining derived database with focus on extracting and classifying gene-disease associations with respect to several biomolecular conditions. It uses a machine learning based algorithm to extract semantic gene-disease relations from a textual source of interest. It is part of the Linked Life Data, of the LMU in Munchen, Germany.[5]
BeFree Data
Extracts gene-disease associations from MEDLINE abstract using the BeFree system. BeFree is composed of a biomedical Named Entity Recognition (BioNER) module to detect diseases and genes and a relation extraction module based on morphosyntactic information.[21]
2.4. Integrative Databases
This sort of databases include Mendelian, compound and environmental diseases in an integrated gene-disease association archive and show that the concept of modularity applies for all of them They provide a functional analysis of diseases in case of important new biological insights, which might not be discovered when considering each of the gene-disease associations independently. Hence, they present a suitable framework for the study of how genetic and environmental factors, such as drugs, contribute to diseases. The best example for this sort of database is DisGeNET.[7][22]
The Gene Disease Associations Database DisGeNET
DisGeNET is a comprehensive gene-disease association database that integrates associations from several sources that covers different biomedical aspects of diseases.[22] In particular, it is focused on the current knowledge of human genetic diseases including Mendelian, complex and environmental diseases. To assess the concept of modularity of human diseases, this database performs a systematic study of the emergent properties of human gene-disease networks by means of network topology and functional annotation analysis.[5] The results indicate a highly shared genetic origin of human diseases and show that for most diseases, including Mendelian, complex and environmental diseases, functional modules exist. Moreover, a core set of biological pathways is found to be associated with most human diseases. Obtaining similar results when studying clusters of diseases, the findings in this database suggest that related diseases might arise due to dysfunction of common biological processes in the cell. The network analysis of this integrated database points out that data integration is needed to obtain a comprehensive view of the genetic landscape of human diseases and that the genetic origin of complex diseases is much more common than expected.[5]
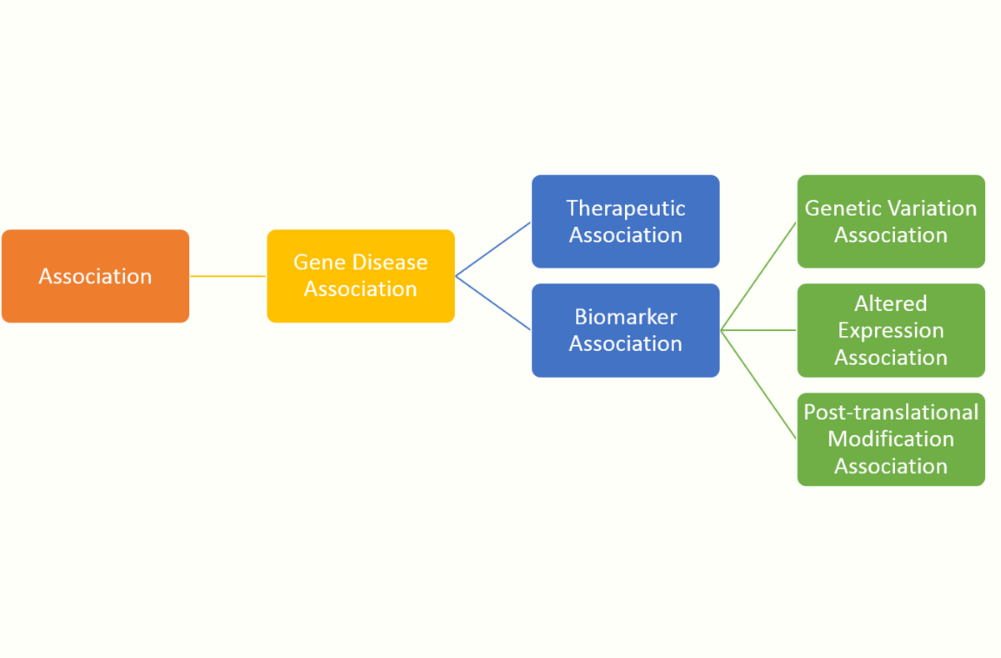
The description of each association type in this ontology is: #Therapeutic Association: The gene/protein has a therapeutic role in the amelioration of the disease. #Biomarker Association: The gene/protein either plays a role in the etiology of the disease (e.g. participates in the molecular mechanism that leads to disease) or is a biomarker for a disease. #Genetic Variation Association: Used when a sequence variation (a mutation, a SNP) is associated to the disease phenotype, but there is still no evidence to say that the variation causes the disease. In some cases the presence of the variants increase the susceptibility to the disease. In general, the NCBI SNP identifiers are provided. #Altered Expression Association: Alterations in the function of the protein by means of altered expression of the gene are associated with the disease phenotype. #Post-translational Modification Association: Alterations in the function of the protein by means of post-translational modifications (methylation or phosphorylation of the protein) are associated with the disease phenotype. [5]
3. Some Use Cases
Some of the most interesting cases using Gene-Disease Databases can be found in the following papers:[5][7]
- Santiago, Jose A.; Potashkin, Judith A. (2014). "A network approach to clinical intervention in neurodegenerative diseases". Trends in Molecular Medicine 20 (12): 694–703. doi:10.1016/j.molmed.2014.10.002. PMID 25455073.
- Kaikkonen, Minna U.; Niskanen, Henri; Romanoski, Casey E.; Kansanen, Emilia; Kivelä, Annukka M.; Laitalainen, Jarkko; Heinz, Sven; Benner, Christopher et al. (2014). "Control of VEGF-A transcriptional programs by pausing and genomic compartmentalization". Nucleic Acids Research 42 (20): 12570–12584. doi:10.1093/nar/gku1036. PMID 25352550.
- Grosdidier, Solène; Ferrer, Antoni; Faner, Rosa; Piñero, Janet; Roca, Josep; Cosío, Borja; Agustí, Alvar; Gea, Joaquim et al. (2014). "Network medicine analysis of COPD multimorbidities". Respiratory Research 15: 111. doi:10.1186/s12931-014-0111-4. PMID 25248857.
- Cristiano, Francesca; Veltri, Pierangelo (2014). "An R-based tool for miRNA data analysis and correlation with clinical ontologies". Proceedings of the 5th ACM Conference on Bioinformatics, Computational Biology, and Health Informatics - BCB '14. pp. 768–773. doi:10.1145/2649387.2660847. ISBN 9781450328944.
- Gallagher, Suzanne Renick; Dombrower, Micah; Goldberg, Debra S. (2014). "Using 2-node hypergraph clustering coefficients to analyze disease-gene networks". Proceedings of the 5th ACM Conference on Bioinformatics, Computational Biology, and Health Informatics - BCB '14. pp. 647–648. doi:10.1145/2649387.2660817. ISBN 9781450328944.
- Mannil, Deepthi; Vogt, Ingo; Prinz, Jeanette; Campillos, Monica (2015). "Organ system heterogeneity DB: A database for the visualization of phenotypes at the organ system level". Nucleic Acids Research 43 (Database issue): D900–D906. doi:10.1093/nar/gku948. PMID 25313158.
- Vogt, Ingo; Prinz, Jeanette; Campillos, Mónica (2014). "Molecularly and clinically related drugs and diseases are enriched in phenotypically similar drug-disease pairs". Genome Medicine 6 (7). doi:10.1186/s13073-014-0052-z.
- Santiago, Jose A.; Potashkin, Judith A. (2014). "System-based approaches to decode the molecular links in Parkinson's disease and diabetes". Neurobiology of Disease 72: 84–91. doi:10.1016/j.nbd.2014.03.019. PMID 24718034.
- Lee, In-Hee; Lee, Kyungjoon; Hsing, Michael; Choe, Yongjoon; Park, Jin-Ho; Kim, Shu Hee; Bohn, Justin M.; Neu, Matthew B. et al. (2014). "Prioritizing Disease-Linked Variants, Genes, and Pathways with an Interactive Whole-Genome Analysis Pipeline". Human Mutation 35 (5): 537–547. doi:10.1002/humu.22520. PMID 24478219.
- Liu, Ming-Xi; Chen, Xing; Chen, Geng; Cui, Qing-Hua; Yan, Gui-Ying (2014). "A Computational Framework to Infer Human Disease-Associated Long Noncoding RNAs". PLOS ONE 9 (1): e84408. doi:10.1371/journal.pone.0084408. PMID 24392133. Bibcode: 2014PLoSO...984408L.
- Zhao, Yilei; Wang, Chen; Wu, Jianwei; Wang, Yan; Zhu, Wenliang; Zhang, Yong; Du, Zhimin (2013). "Choline Protects Against Cardiac Hypertrophy Induced by Increased After-load". International Journal of Biological Sciences 9 (3): 295–302. doi:10.7150/ijbs.5976. PMID 23493786.
- Koczor, Christopher A.; Lee, Eva K.; Torres, Rebecca A.; Boyd, Amy; Vega, J. David; Uppal, Karan; Yuan, Fan; Fields, Earl J. et al. (2013). "Detection of differentially methylated gene promoters in failing and nonfailing human left ventricle myocardium using computation analysis". Physiological Genomics 45 (14): 597–605. doi:10.1152/physiolgenomics.00013.2013. PMID 23695888.
- Gu, Ying; Liu, Guang-Hui; Plongthongkum, Nongluk; Benner, Christopher; Yi, Fei; Qu, Jing; Suzuki, Keiichiro; Yang, Jiping et al. (2014). "Global DNA methylation and transcriptional analyses of human ESC-derived cardiomyocytes". Protein & Cell 5: 59–68. doi:10.1007/s13238-013-0016-x. PMID 24474197.
- Galhardo, Mafalda; Sinkkonen, Lasse; Berninger, Philipp; Lin, Jake; Sauter, Thomas; Heinäniemi, Merja (2014). "Integrated analysis of transcript-level regulation of metabolism reveals disease-relevant nodes of the human metabolic network". Nucleic Acids Research 42 (3): 1474–1496. doi:10.1093/nar/gkt989. PMID 24198249.
- Tieri, Paolo; Termanini, Alberto; Bellavista, Elena; Salvioli, Stefano; Capri, Miriam; Franceschi, Claudio (2012). "Charting the NF-κB Pathway Interactome Map". PLOS ONE 7 (3): e32678. doi:10.1371/journal.pone.0032678. PMID 22403694. Bibcode: 2012PLoSO...732678T.
4. Remarks about the Future in Gene Disease Databases
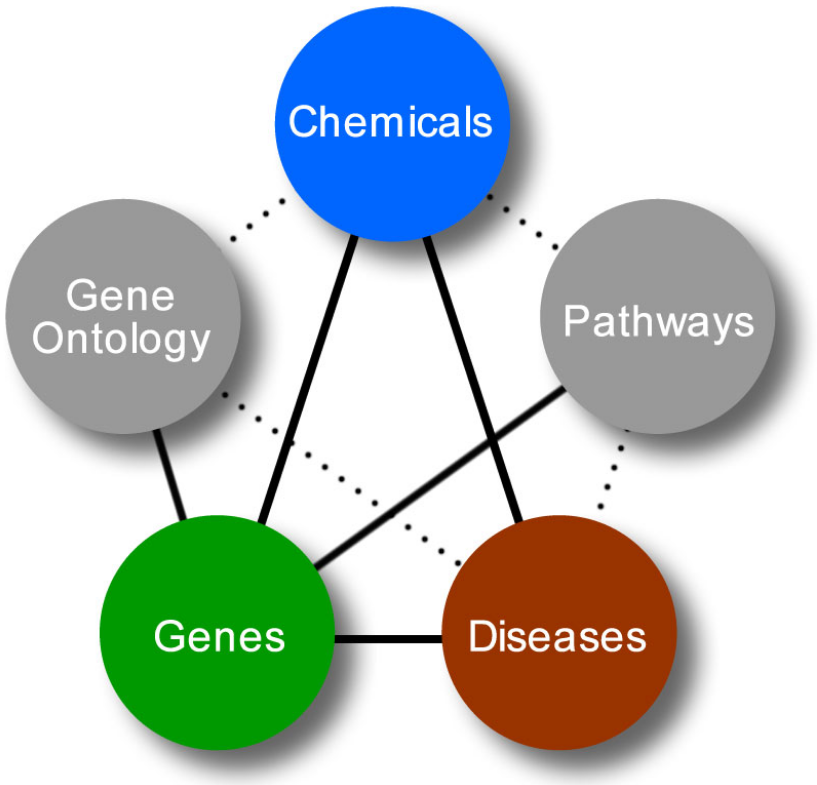
The completion of the human genome has changed the way the search for disease genes is performed. In the past, the approach was to focus on one or a few genes at a time. Now, projects like the DisGeNET exemplify the efforts to systematically analyze all the gene alterations involved in a single or multiple diseases.[23] The next step is to produce a complete picture of the mechanistic aspects of the diseases and the design of drugs against them. For that, a combination of two approaches will be needed: a systematic search and in-depth study of each gene. The future of the field will be defined by new techniques to integrate large bodies of data from different sources and to incorporate functional information into the analysis of large-scale data generated by bioinformatics studies.[5]
Bioinformatics is both a term for the body of biological gene disease studies that use computer programming as part of their methodology, as well as a reference to specific analysis pipelines that are repeatedly used, particularly in the fields of genetics and genomics.[5] Common uses of bioinformatics include the identification of candidate genes and nucleotides, SNPs. Often, such identification is made with the aim of better understanding the genetic basis of disease, unique adaptations, desirable properties, or differences between populations. In a less formal way, bioinformatics also tries to understand the organisational principles within nucleic acid and protein sequences.[5]
The response of bioinformatics to new experimental techniques brings a new perspective into the analysis of the experimental data, as demonstrated by the advances in the analysis of information from gene disease databases and other technologies. It is expected that this trend will continue with novel approaches to respond to new techniques, such as next-generation sequencing technologies. For instance, the availability of large numbers of individual human genomes will promote the development of computational analyses of rare variants, including the statistical mining of their relations to lifestyles, drug interactions and other factors.[5] Biomedical research will also be driven by our ability to efficiently mine the large body of existing and continuously generated biomedical data. Text-mining techniques, in particular, when combined with other molecular data, can provide information about gene mutations and interactions and will become crucial to stay ahead of the exponential growth of data generated in biomedical research. Another field that is benefiting from the advances in mining and integration of molecular, clinical and drug analysis is pharmacogenomics. In silico studies of the relationships between human variations and their effect on diseases will be key to the development of personalized medicine.[7] In summary, Gene Disease Databases have already transformed the search for disease genes and has the potential to become a crucial component of other areas of medical research.[5]
The content is sourced from: https://handwiki.org/wiki/Biology:Gene_Disease_Database
References
- American Medical Informatics Association, "American Medical Informatics Association Strategic Plan," August 2011. [Online]. Available: http://www.amia.org/inside/stratplan/. [Accessed 15 October 2014].
- Botstein, D (2003). "Discovering genotypes underlying human phenotypes: past successes for Mendelian disease, future approaches for complex disease". Nature Genetics 33 (1): 228–237. doi:10.1038/ng1090. PMID 12610532. https://dx.doi.org/10.1038%2Fng1090
- Oti, M (2007). "The modular nature of genetic diseases". Clinical Genetics 71 (1): 1–11. doi:10.1111/j.1399-0004.2006.00708.x. PMID 17204041. https://dx.doi.org/10.1111%2Fj.1399-0004.2006.00708.x
- Davis, A.; King, B. (2011). "The Comparative Toxicogenomics Database: update 2011". Nucleic Acids Res. 39 (1): 1067–1072. doi:10.1093/nar/gkq813. PMID 20864448. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3013756
- A. Bauer-Mehren, "Gene-Disease network Analysis Reveals Functional Modules in Mendelian, Complex and Environmental diseases," PLOS ONE, pp. 1-3, 2011.
- Davis, A.; Wiegers, T. (2013). "Text Mining Effectively Scores and Ranks the Literature for Improving Chemical-Gene-Disease Curation at the Comparative Toxicogenomics Database". PLoS ONE 8 (4): 1–29. doi:10.1371/journal.pone.0058201. PMID 23613709. Bibcode: 2013PLoSO...858201D. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3629079
- Bauer-Mehren, A.; Rautscha, M. (2010). "DisGeNET: a Cytoscape plugin to visualize, integrate, search and analyze gene–disease networks". Bioinformatics 26 (22): 2924–2926. doi:10.1093/bioinformatics/btq538. PMID 20861032. https://dx.doi.org/10.1093%2Fbioinformatics%2Fbtq538
- Vogt, I. (2014). "Systematic analysis of gene properties influencing organ system phenotypes in mammalian perturbations". Bioinformatics 30 (21): 3093–3100. doi:10.1093/bioinformatics/btu487. https://dx.doi.org/10.1093%2Fbioinformatics%2Fbtu487
- Buneman, P. (2008). "Curated Databases". Bibliometrics 978 (1): 152–162.
- Murphy, C.; Davis, A. (2009). "Comparative Toxicogenomics Database: a knowledgebase and discovery tool for chemical–gene–disease networks". Bioinformatics 37 (1): 786–792. doi:10.1093/nar/gkn580. PMID 18782832. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2686584
- "The Universal Protein Resource (UniProt)". Nucleic Acids Research 36 (1): 190–195. 2008. doi:10.1093/nar/gkm895. PMID 18045787. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1669721
- K. Brown, "Online Predicted human Interaction Database," Bioinformatics, vol. 21, no. 9, pp. 2076-2082, 2005.
- S. Hunter and P. Jones, "InterPro in 2011: new developments in the family and domain prediction database," Nucleic Acids Research, vol. 10, no. 1, pp. 12-22, 2011
- C. Bult and J. Eppig, "The Mouse genome Database (MGD): mouse biology and model systems," Nucleic Acids Research, vol. 36, no. 1, pp. 724-728, 2007
- M. Dwinell, E. Worthey and S. M, "The Rat genome Database 2009: variation, ontologies and pathways," Nucleic Acids Research, vol. 37, no. 1, pp. 744-749, 2009
- "The Rat Genome Database 2015: genomic, phenotypic and environmental variations and disease". Nucleic Acids Research 43 (Database issue): D743–50. 2015. doi:10.1093/nar/gku1026. PMID 25355511. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4383884
- A. Homosh, "Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders," Nucleic Acids Research, vol. 33, no. 1, pp. 514-517, 2005
- P. Flicek and M. Ridwan, "Ensembl 2012," Nucleic Acids Research, vol. 40, no. 1, pp. 84-90, 2012
- Becker, K.; Barnes, K. (2004). "The genetic Association Database". Nature Genetics 36 (5): 431–432. doi:10.1038/ng0504-431. PMID 15118671. https://dx.doi.org/10.1038%2Fng0504-431
- https://geneticassociationdb.nih.gov/
- Bravo, A (2014). "Extraction of relations between genes and diseases from text and large-scale data analysis: implications for translational research". BMC Bioinformatics 16 (1): 55. doi:10.1186/s12859-015-0472-9. PMID 25886734. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4466840
- Piñero (2015). "DisGeNET: a discovery platform for the dynamical exploration of human diseases and their genes". Database 2015: bav028. doi:10.1093/database/bav028. PMID 25877637. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4397996
- Oti, M (2006). "Predicting disease genes using protein-protein interactions". J. Med. Genet. 43 (8): 691–698. doi:10.1136/jmg.2006.041376. PMID 16611749. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2564594