Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
The physical interactions between enhancers and promoters create chromatin conformations involved in gene regulation. Although it is not entirely comprehensive how chromatin-mediated enhancer–promoter (E–P) interactions with various histone marks can affect gene expression, this proximity has been observed in multiple systems at multiple loci and is thought to be essential to control gene expression.
- gene expression
- enhancer
- promoter
- histone modifications
1. Introduction
The spatial organization of the genome into transcriptionally active and silenced chromatin plays a fundamental role in the three-dimensional (3D) architecture required for the regulation of eukaryotic genes [1,2]. Genomic sequences are partitioned into one of two nuclear compartments called the A/B compartments: the A compartment is an ‘open’ state that allows for the transcription of the associated genes (euchromatin), and the B compartment is a ‘closed’ state associated with inactive genes (heterochromatin) [3]. The compacted chromatin of heterochromatin is assumed to be inaccessible to transcriptional machinery and resistant to chromatin remodeling; this condensed state is thus accepted as a major hallmark of repressed chromatin, which comprises silenced genes [4]. Heterochromatin is further divided into two types, constitutive and facultative. In constitutive heterochromatin, repetitive sequences such as pericentromeric regions are organized into silent nuclear compartments that are highly enriched in trimethylated histone H3 lysine 9 (H3K9me3) and methylated DNA [5]. In contrast, facultative heterochromatin consists of transcriptionally silent regions that become activated depending on the context [6].
In one key mechanism, gene transcription is regulated through chromatin loops that form between promoters and various regulatory elements, including enhancers [3,7]. Enhancers are cis-elements that contain diverse DNA sequences to which various transcription factors (TFs) and transcriptional co-activators bind and that are enriched with various histone modifications that facilitate gene transcription [8,9,10].
2. Histone Modifications Involved in E–P Interactions
Histone modification is widely used as a means to classify enhancers according to their activity: H3K4me1 and the binding of trithorax-related mixed lineage leukemia (MLL) complex define primed or active enhancers; H3K27me3 is a key marker of poised or inactive enhancers; histone H3 lysine 27 acetylation (H3K27ac) is a hallmark of transcriptionally active enhancers (Figure 1) [11,12,13,14]. Recent trends highlight that rather than defining active enhancers with H3K27ac, different histone acetylation marks, such as simultaneous acetylation of histone H4 at both K5 and K8 (H4K5acK8ac) [15], Das et al., [submitted], histone H2B N-terminus multisite lysine (e.g., K5, K12, K16, and K20) acetylation (H2BNTac [16]), H3K122ac [17], and H4K16ac [18], to define active enhancers are emerging. A large number of histone modifications have been implicated in gene transcription, where H3K4me3 has been associated with gene promoter regions [19]. Table 1 summarizes the most well-characterized histone modifications and their involvement in E–P interactions.
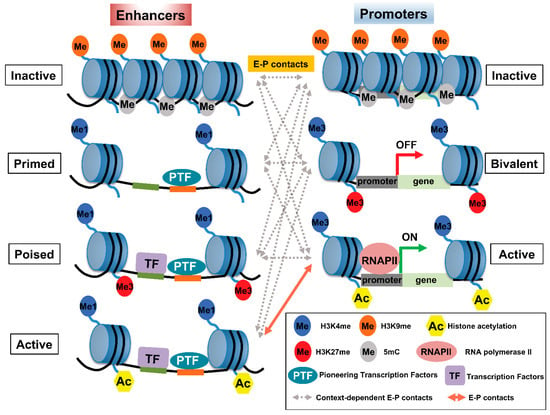
Figure 1. Pattern of histone modifications defines enhancer–promoter (E–P) states. (Left panel) The pattern of histone modifications in various enhancer states. Inactive enhancers are characterized by high nucleosome density and enrichment of H3K9me2/3 and DNA methylation. At primed enhancers, pioneering TFs (PTF) bind to their target sites and recruit MLL3/4 complexes to decorate H3K4me1. PTF binding is accompanied by nucleosome remodeling. H3K27me3 may contribute to the poised status of an enhancer to prevent premature activation. In addition, poised enhancers may allow lineage-specific TFs (LTF) to bind and recruit histone acetyltransferases, such as p300/CBP, and H3K27 demethylase(s) to prepare the enhancer for rapid activation. Active enhancers contain not only H3K27ac—the hallmark of an active enhancer—but also H4K5acK8ac, H2BNTac, and potentially acetylation at various residues of other histones. (Right panel) The pattern of histone modifications in various promoter states. Promoter activity is highly correlated with enhancer activity and the associated epigenetic landscape. Inactive promoters carry high levels of H3K9me2/3 and DNA methylation. The vast majority of each promoter is marked by H3K4me3, whereas other parts of the promoters are marked by H3K27me3, thus constituting so-called ‘bivalent promoters,’ which are repressed by polycomb group complexes. In comparison, active promoters gain histone acetylation marks. The epigenetic landscape of enhancers and promoters can be variably altered in response to external signals. However, the timing of and how E–P interactions facilitate gene expression is not well understood and may involve context-dependent interactions among enhancers and promoters at various states.
Hyperacetylation of histone H4 at its N-terminal tail is essential for normal spermatogenesis [20,21] and occurs in several cancers, including nuclear protein in testis (NUT) midline carcinoma [22]. The bromodomain (BRD) and extra-terminal domain (BET) proteins, including BRD4, preferentially bind to histone H4 tails containing multiple acetylations within 1 to 5 amino acids, e.g., H4K5acK8ac, and only rarely bind to mono-acetylated histone species, including H3K27ac [23,24,25]. Although BRD4 primarily binds to multi-acetylated histone H4, most previous studies of BRD4 have focused on H3K27ac. BET proteins, including BRD4, have long been associated with large-scale control of the nuclear structure and higher-order chromatin organization [26,27]. For example, BRD4-NUT is an oncogenic fusion protein that drives NUT carcinoma [28]. NUT is normally expressed in post-meiotic spermatogenic cells, where it interacts with p300 and triggers genome-wide histone hyperacetylation [21]. Such interactions among BRD4-NUT, p300, and histone hyperacetylation lead to the formation of hyperacetylated nuclear ‘foci,’ corresponding to large chromosomal ‘megadomains’ (100 kb to 2 Mb) [22], which involve long-range acetylation-dependent inter- and intrachromosomal interactions [29]. This example illustrates how histone hyperacetylation (e.g., H4K5acK8ac, H2BNTac), targeted by readers or writers of BRD-containing proteins, facilitates long-range contact between enhancers and promoters and controls gene expression and how dysregulation of this process leads to disease, including cancer.
Table 1. Summary of histone modifications and their putative role in enhancer–promoter (E–P) interactions and transcription.
| Histone Modification | Putative Role in E–P Interactions and Transcription | Most Enriched Region | Reference |
|---|---|---|---|
| H3K4me3 | Activation | Promoters, bivalent promoters | [19] |
| H3K4me1 | Activation | Enhancers | [14] |
| H3K27ac | Activation | Enhancers, promoters | [11,12,13,14] |
| H4K5acK8ac | Activation | Enhancers, promoters | [15] |
| H4K16ac | Activation | Enhancers | [18] |
| H2BNTac (K5, K12, K16, K20) | Activation | Enhancers | [16] |
| H3K27me3 | Repression | Bivalent Promoters, poised enhancers | [30,31,32,33,34] |
H3K27me3 is present at high levels in CpG islands, which are associated with the promoters of developmental genes in mammals [30,31]. In addition, along with H3K4me1, H3K27me3 is plentiful in poised enhancers (PEs), which become activated in human and mouse embryonic stem cells once these PEs are ‘marked’ with acetylation at H3K27 during the activation of associated genes [14]. In most cases, PEs are linked to developmental genes that are inactive in embryonic stem cells (ESCs) and that are expressed upon differentiation. Therefore, the PE chromatin signature is proposed to bookmark associated genes spatiotemporally and facilitate their activation once appropriate differentiation signals arise (Figure 1). Accordingly, loss of polycomb repressive complex (PRC) 2, which catalyzes the methylation of H3K27, induces a genome-wide redistribution of H3K27ac marking and activation of PEs [32]. The functional role of the polycomb-repressed or -poised state of enhancers remains unknown. Several studies suggest that polycomb-repressed enhancers may convert these regions to silencers [33,34], whereas PEs may be associated with the rapid activation of genes [35].
How do specific enhancers locate and contact promoters to accomplish transcriptional regulation? Remarkable progress has been achieved regarding the molecular principles and combinatorial logic underlying these processes [3,7], but chromatin-mediated E–P interactions remain incompletely understood. In general, chromosomal interactions create microenvironments, such as topologically associating domains (TADs, highly-interacting genomic regions defined as the population-level contact-frequency domains of higher interaction frequency within a region than between regions, [36]) and nuclear compartments that are characterized by the clustering of similar epigenetic marks, which constrain the search space and thereby increase the likelihood of interactions between enhancers and promoters in the genome [37]. In one model [7], interdependent layers of regulatory control cooperate to establish and maintain E–P interactions for robust cell-type–specific gene expression. Transcriptionally favorable cis-regulatory elements contain TFs, RNA polymerase II, and chromatin-remodeling and histone-modifying enzymes, whose combined activities induce chromatin accessibility. Specifically, proteins bound to enhancers and promoters may interact with each other non-randomly and preferentially [38,39], leading to cooperations that influence transcriptional regulation [7]. Chromatin loops that are marked with high levels of H3K27ac and low levels of H3K27me3 tend to change upon perturbation of PRC2, thus providing evidence that histone modification can alter the overall genomic architecture [40]. In cancer cells, H3K27ac dynamics modulate the interaction frequency between regulatory regions and can lead to allele-specific chromatin configurations that sustain oncogene expression [41].
This entry is adapted from the peer-reviewed paper 10.3390/cancers14215404
This entry is offline, you can click here to edit this entry!