Gene therapy using the adeno-associated virus (rAAV) to deliver mini/micro- dystrophin is the current promising strategy for Duchenne Muscular Dystrophy (DMD). However, the further transformation of this strategy still faces many “bottlenecks”. Most gene therapies are only suitable for infants with strong muscle cell regeneration and immature immune system, and the treatment depends heavily on the high dose of rAAV. However, high-dose rAAV inevitably causes side effects such as immune response and acute liver toxicity. Therefore, how to reduce the degree of fibrosis and excessive immune response in older patients and uncouple the dependence association between therapeutic effect and high dose rAAV are crucial steps for the transformation of rAAV-based gene therapy.
- Ducheen muscular dystrophin
- gene therapy
- MyoAAV
1. Introduction
2. Clinical Development of Systemic AAV-Mediated Mini/Micro-Dystrophin Gene Therapy
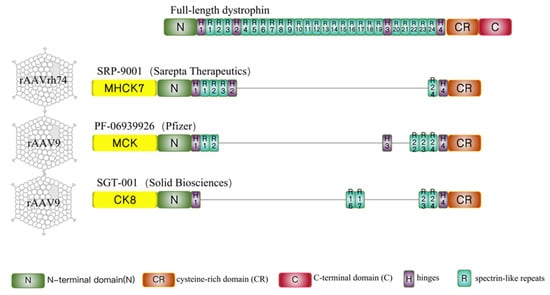
| Sarepta | Pfizer | Therapeutics Solid Biosciences | |
|---|---|---|---|
| Trial name | An open-label, systemic gene delivery study using commercial process material to evaluate the safety of and expression from SRP-9001 in subjects with Duchenne muscular dystrophy (ENDEAVOR) | A phase 1b multicenter, open-label, single ascending dose study to evaluate the safety and tolerability of pf-06939926 in ambulatory and non-ambulatory subjects with Duchenne muscular dystrophy | A randomized, controlled, open-label, single-ascending dose, phase I/II study to investigate the safety and tolerability, and efficacy of intravenous SGT-001 in male adolescents and children with Duchenne muscular dystrophy |
| ClinicalTrials.gov Identififier | NCT04626674 | NCT03362502 | NCT03368742 |
| Study nature | Phase-1b trial | Phase 1b, open-label, trial | Phase 1/2, open-label, trial |
| Drug name | SRP-9001 | PF-06939926 | SGT-001 |
| AAV-serotype | rAAV-rh74 | rAAV9 | rAAV9 |
| Dose | 1 dose (1.33 × 1014 vg/kg) for cohort 1 | 2 doses (1.0 × 1014 vg/kg, 3.0 × 1014 vg/kg) | 2 doses (5.0 × 1013 vg/kg 2.0 × 1014 vg/kg) |
| Patient number | 38 | 22 | 16 (estimated enrollment) |
| Patient average age | 3 years and older | 4 years and older | 4~17 years |
| Disease stage | Ambulatory and non-ambulatory subjects | Ambulatory and non-ambulatory subjects | Ambulatory and non-ambulatory subjects |
| Corticosteroid use | 3 months on stable weekly dose of oral corticosteroids for cohort 1 | Daily glucocorticoids for at least 3 months | Stable daily dose (or equivalent) of oral corticosteroids ≥ 12 wks |
| Dystrophin gene mutation | Any mutation | Any mutation | Any mutation |
| Pre-Nab to AAV | Negative | Negative | Negative |
| Primary outcome | The change in micro-dystrophin expression in DMD patients treated with SRP-9001. | Safety and tolerability | Safety |
| Secondary outcome | Adverse events, vector shedding, and the development of antibodies to AAVrh74. | Micro-dystrophin expression in biopsy | |
| SAE | Increased transaminases that required corticosteroid treatment. Nausea and vomiting that required intravenous treatment (cohort 1). |
More than 40% of patients suffered vomiting, nausea, decreased appetite, and pyrexia. | Complement activation, reduced platelet count, liver dysfunction, and acute kidney injury |
2.1. Sarepta-SRP-9001
2.2. Pfizer-PF-06939926
2.3. Solid Biosciences-SGT-001
References
- Moser, H. Duchenne muscular dystrophy: Pathogenetic aspects and genetic prevention. Hum. Genet. 1984, 66, 17–40.
- Birnkrant, D.J.; Panitch, H.B.; Benditt, J.O.; Boitano, L.J.; Carter, E.R.; Cwik, V.A.; Finder, J.D.; Iannaccone, S.T.; Jacobson, L.E.; Kohn, G.L.; et al. American College of Chest Physicians consensus statement on the respiratory and related management of patients with Duchenne muscular dystrophy undergoing anesthesia or sedation. Chest 2007, 132, 1977–1986.
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517.
- Ibraghimov-Beskrovnaya, O.; Ervasti, J.M.; Leveille, C.J.; Slaughter, C.A.; Sernett, S.W.; Campbell, K.P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 1992, 355, 696–702.
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714.
- McDonald, C.M.; Henricson, E.K.; Abresch, R.T.; Han, J.J.; Escolar, D.M.; Florence, J.M.; Duong, T.; Arrieta, A.; Clemens, P.R.; Hoffman, E.P.; et al. The cooperative international neuromuscular research group Duchenne natural history study—A longitudinal investigation in the era of glucocorticoid therapy: Design of protocol and the methods used. Muscle Nerve 2013, 48, 32–54.
- Campbell, K.P. Three muscular dystrophies: Loss of cytoskeleton-extracellular matrix linkage. Cell 1995, 80, 675–679.
- Naso, M.F.; Tomkowicz, B.; Perry, W.L.; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334.
- Sonntag, F.; Köther, K.; Schmidt, K.; Weghofer, M.; Raupp, C.; Nieto, K.; Kuck, A.; Gerlach, B.; Böttcher, B.; Müller, O.J.; et al. The assembly-activating protein promotes capsid assembly of different adeno-associated virus serotypes. J. Virol. 2011, 85, 12686–12697.
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug. Discov. 2019, 18, 358–378.
- England, S.B.; Nicholson, L.V.; Johnson, M.A.; Forrest, S.M.; Love, D.R.; Zubrzycka-Gaarn, E.E.; Bulman, D.E.; Harris, J.B.; Davies, K.E. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature 1990, 343, 180–182.
- Wilton-Clark, H.; Yokota, T. Antisense and Gene Therapy Options for Duchenne Muscular Dystrophy Arising from Mutations in the N-Terminal Hotspot. Genes 2022, 13, 257.
- Salva, M.Z.; Himeda, C.L.; Tai, P.W.; Nishiuchi, E.; Gregorevic, P.; Allen, J.M.; Finn, E.E.; Nguyen, Q.G.; Blankinship, M.J.; Meuse, L.; et al. Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol. Ther. 2007, 15, 320–329.
- Mendell, J.R.; Sahenk, Z.; Lehman, K.; Nease, C.; Lowes, L.P.; Miller, N.F.; Iammarino, M.A.; Alfano, L.N.; Nicholl, A.; Al-Zaidy, S.; et al. Assessment of Systemic Delivery of rAAVrh74.MHCK7.micro-dystrophin in Children With Duchenne Muscular Dystrophy: A Nonrandomized Controlled Trial. J. AMA. Neurol. 2020, 77, 1122–1131.
- Sarepta Therapeutics, I. Sarepta Therapeutics’ Investigational Gene Therapy SRP-9001 for Duchenne Muscular Dystrophy Demonstrates Significant Functional Improvements Across Multiple Studies. Available online: https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics-investigational-gene-therapy-srp-9001 (accessed on 12 October 2022).
- Sarepta Therapeutics, I. Sarepta Therapeutics Announces Top-Line Results for Part 1 of Study 102 Evaluating SRP-9001, Its Investigational Gene Therapy for the Treatment of Duchenne Muscular Dystrophy. Available online: https://investorrelations.Sarepta.com/news-releases/news-release-details/Sarepta-therapeutics-announces-top-line-results-part-1-study-102 (accessed on 5 October 2022).
- Clinicaltrials.gov. A Gene Transfer Therapy Study to Evaluate the Safety of and Expression from SRP-9001 in Participants with Duchenne Muscular Dystrophy (DMD). Available online: https://clinicaltrials.gov/ct2/show/NCT04626674 (accessed on 5 October 2022).
- Zaidman, C.P.C.; Mcdonald, C.; Giblin, K.; Collins, L.; Wang, S.; Upadhyay, S.; Lewis, S.; Malhotra, J.; Griffin, D.A. ENDEAVOR: A Gene Delivery Study to Evaluate the Safety of and Expression from SRP-9001 in Duchenne Muscular Dystrophy. Available online: https://investorrelations.sarepta.com/static-files/a674d68e-823c-43a4-b26c-e6bfc6a5a95b (accessed on 7 October 2022).
- Sarepta Therapeutics, I. Sarepta Therapeutics’ SRP-9001 Shows Sustained Functional Improvements in Multiple Studies of Patients with Duchenne. Available online: https://investorrelations.Sarepta.com/news-releases/news-release-details/Sareptatherapeutics-SRP-9001-shows-sustained-functional (accessed on 7 October 2022).
- Clinicaltrials.gov. A Gene Transfer Therapy Study to Evaluate the Safety and Efficacy of SRP-9001 in Participants with Duchenne Muscular Dystrophy (DMD). Available online: https://clinicaltrials.gov/ct2/show/NCT05096221 (accessed on 5 October 2022).
- Clinicaltrials.gov. Pfizer’s New Phase 1b Results of Gene Therapy in Ambulatory Boys with Duchenne Muscular Dystrophy (DMD) Support Advancement into Pivotal Phase 3 Study. Available online: https://investors.pfizer.com/investor-news/press-releasedetails/2020/Pfizers-New-Phase-1b-Results-of-Gene-Therapy-in-Ambulatory-Boys-with-Duchenne-Muscular-DystrophyDMD-Support-Advancement-into-Pivotal-Phase-3-Study/default.aspx (accessed on 27 October 2022).
- Wang, B.; Li, J.; Fu, F.H.; Chen, C.; Zhu, X.; Zhou, L.; Jiang, X.; Xiao, X. Construction and analysis of compact muscle-specific promoters for AAV vectors. Gene Ther. 2008, 15, 1489–1499.
- Safety and Efficacy of pf-06939926 Gene Therapy in Boys with Duchenne Muscular Dystrophy: Update on Data from the Phase 1b Study | Mda Clinical & Scientific Conference 2022. Available online: https://mdaconference.org/index.php/node/1168 (accessed on 9 December 2021).
- Clinicaltrials.gov. A Phase 3 Study to Evaluate the Safety and Efficacy of pf-06939926 for the Treatment of Duchenne Muscular Dystrophy. Available online: https://clinicaltrials.gov/ct2/show/NCT04281485 (accessed on 5 October 2022).
- Biotech, F. Pfizer Tightens DMD Patient Criteria after Serious Adverse Events Crop up in Phase 3 Gene Therapy Trial | Fiercebiotech. Available online: https://www.fiercebiotech.com/biotech/pfizer-tightening-Duchenne-muscular-dystrophy-phase-3-criteriaadverse-event/ (accessed on 5 October 2022).
- Biotech, F. Pfizer Reports Patient Death in Early-Stage Duchenne Gene Therapy Trial, Halts Enrollment. Available online: https://www.fiercebiotech.com/biotech/pfizer-reports-death-patient-Duchenne-trial-halts-enrolment (accessed on 5 October 2022).
- Duan, D. Systemic AAV Micro-dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2018, 26, 2337–2356.
- BioSpace. FDA Slaps Clinical Hold on Solid Bioscience DMD Gene Therapy Program. Available online: https://www.biospace.com/article/fda-slaps-clinical-hold-on-solid-bioscience-DMD-gene-therapy-program/ (accessed on 6 October 2022).
- BioSpace. FDA Slaps Second Clinical Hold on Solid Biosciences’ DMD Gene Therapy Due to Adverse Event. Available online: https://www.biospace.com/article/fda-slaps-second-clinical-hold-on-solid-biosciences-DMD-gene-therapy-due-toadverse-events/ (accessed on 7 October 2022).
- Zaiss, A.K.; Cotter, M.J.; White, L.R.; Clark, S.A.; Wong, N.C.W.; Holers, V.M.; Bartlett, J.S.; Muruve, D.A. Complement is an essential component of the immune response to adeno-associated virus vectors. J. Virol. 2008, 82, 2727–2740.