Covalent inhibitors are a class of small molecule compounds that can covalently bind to specific target proteins, thereby inhibiting their biological functions. Cysteine is one of the least abundant amino acids in proteins of many organisms, which plays a crucial role in catalysis, signal transduction, and redox regulation of gene expression. The thiol group of cysteine possesses the ability to perform nucleophilic and redox-active functions that are not feasible for other natural amino acids. Cysteine is the most common covalent amino acid residue and has been shown to react with a variety of warheads, especially Michael receptors.
- cysteine
- covalent warheads
- covalent inhibitor
1. Introduction
2. Covalent Inhibitors
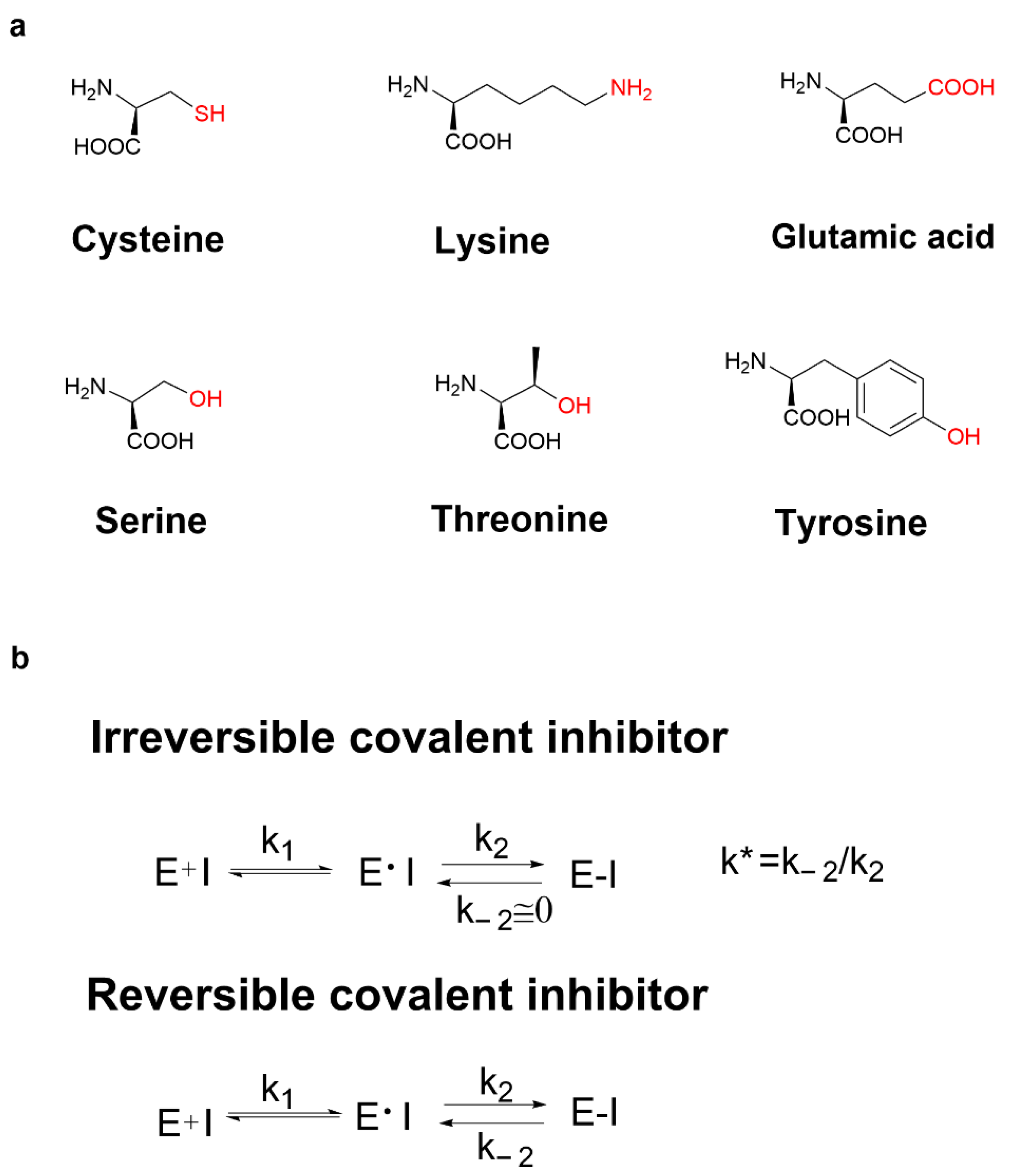
3. Covalent Inhibitors Covalently Bound to Cysteine
3.1. Cysteine Profile
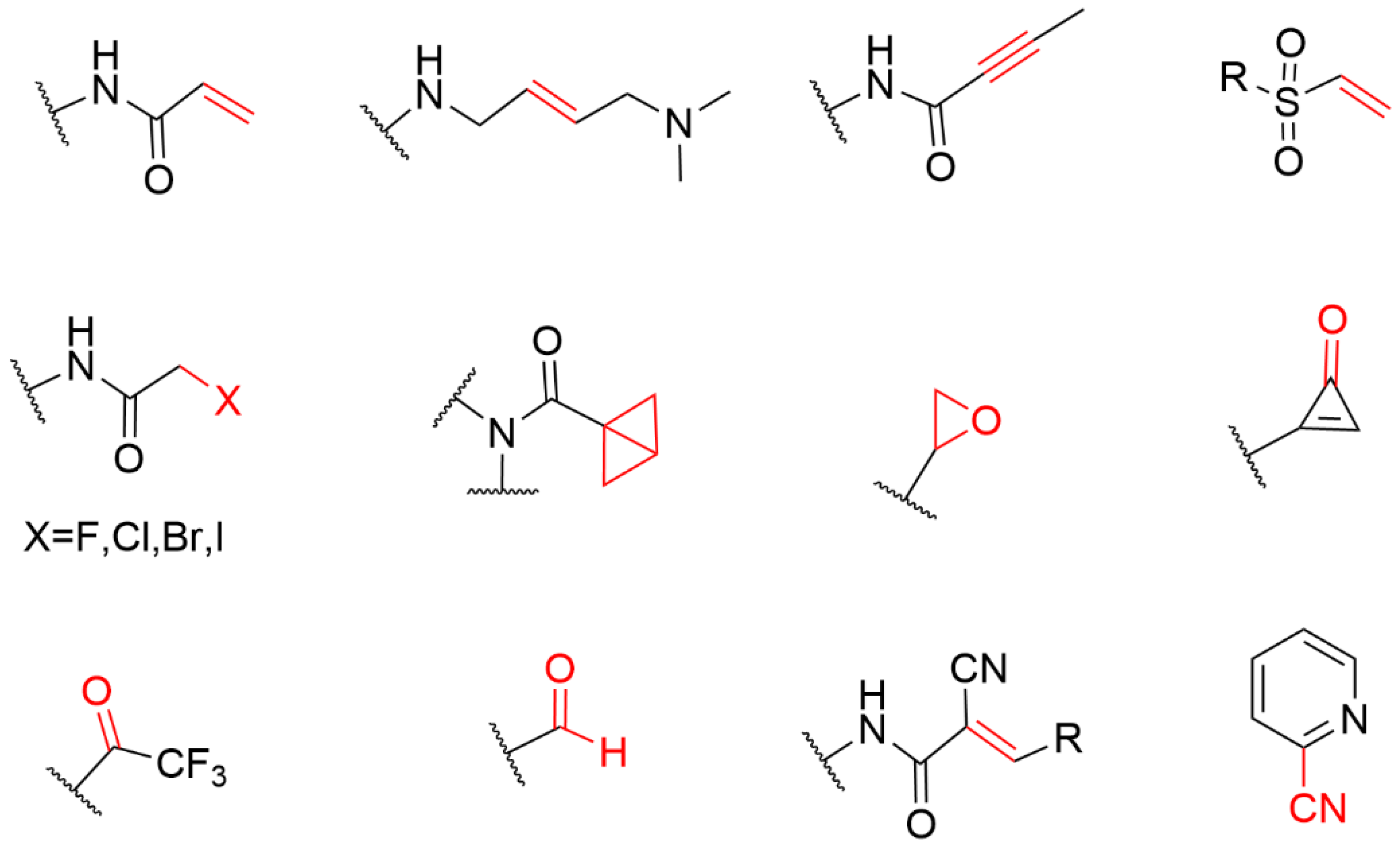
3.2. Cysteine-Directed Covalent Drugs
This entry is adapted from the peer-reviewed paper 10.3390/molecules27227728
References
- Uetrecht, J. Idiosyncratic drug reactions: Past, present, and future. Chem. Res. Toxicol. 2008, 21, 84–92.
- Bradshaw, J.M.; McFarland, J.M.; Paavilainen, V.O.; Bisconte, A.; Tam, D.; Phan, V.T.; Romanov, S.; Finkle, D.; Shu, J.; Patel, V.; et al. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol. 2015, 11, 525–531.
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M.; et al. Clinical Acquired Resistance to KRASG12C Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS–MAPK Reactivation. Cancer Discov. 2021, 11, 1913–1922.
- Jung, Y.; Noda, N.; Takaya, J.; Abo, M.; Toh, K.; Tajiri, K.; Cui, C.; Zhou, L.; Sato, S.-I.; Uesugi, M. Discovery of Non-Cysteine-Targeting Covalent Inhibitors by Activity-Based Proteomic Screening with a Cysteine-Reactive Probe. ACS Chem. Biol. 2022, 17, 340–347.
- Kim, H.-R.; Tagirasa, R.; Yoo, E. Covalent Small Molecule Immunomodulators Targeting the Protease Active Site. J. Med. Chem. 2021, 64, 5291–5322.
- Carrera, A.C.; Alexandrov, K.; Roberts, T.M. The conserved lysine of the catalytic domain of protein kinases is actively involved in the phosphotransfer reaction and not required for anchoring ATP. Proc. Natl. Acad. Sci. USA 1993, 90, 442–446.
- Quach, D.; Tang, G.; Anantharajan, J.; Baburajendran, N.; Poulsen, A.; Wee, J.L.K.; Retna, P.; Li, R.; Liu, B.; Tee, D.H.Y.; et al. Strategic Design of Catalytic Lysine-Targeting Reversible Covalent BCR-ABL Inhibitors. Angew Chem Int Ed Engl 2021, 60, 17131–17137.
- Martín-Gago, P.; Fansa, E.K.; Winzker, M.; Murarka, S.; Janning, P.; Schultz-Fademrecht, C.; Baumann, M.; Wittinghofer, A.; Waldmann, H. Covalent Protein Labeling at Glutamic Acids. Cell Chem. Biol. 2017, 24, 589–597.
- Powers, J.C.; Asgian, J.L.; Ekici, Ö.D.; James, K.E. Irreversible Inhibitors of Serine, Cysteine, and Threonine Proteases. Chem. Rev. 2002, 102, 4639–4750.
- Fadeyi, O.O.; Hoth, L.R.; Choi, C.; Feng, X.; Gopalsamy, A.; Hett, E.C.; Kyne, R.E.; Robinson, R.P.; Jones, L.H. Covalent Enzyme Inhibition through Fluorosulfate Modification of a Noncatalytic Serine Residue. ACS Chem. Biol. 2017, 12, 2015–2020.
- Bender, A.T.; Gardberg, A.; Pereira, A.; Johnson, T.; Wu, Y.; Grenningloh, R.; Head, J.; Morandi, F.; Haselmayer, P.; Liu-Bujalski, L. Ability of Bruton’s Tyrosine Kinase Inhibitors to Sequester Y551 and Prevent Phosphorylation Determines Potency for Inhibition of Fc Receptor but not B-Cell Receptor Signaling. Mol. Pharmacol. 2017, 91, 208–219.
- Gu, C.; Shannon, D.A.; Colby, T.; Wang, Z.; Shabab, M.; Kumari, S.; Villamor, J.G.; McLaughlin, C.J.; Weerapana, E.; Kaiser, M. Chemical Proteomics with Sulfonyl Fluoride Probes Reveals Selective Labeling of Functional Tyrosines in Glutathione Transferases. Chem. Biol. 2013, 20, 541–548.
- Liu, Q.; Sabnis, Y.; Zhao, Z.; Zhang, T.; Buhrlage, S.J.; Jones, L.H.; Gray, N.S. Developing Irreversible Inhibitors of the Protein Kinase Cysteinome. Chem. Biol. 2013, 20, 146–159.
- Pace, N.J.; Weerapana, E. Diverse Functional Roles of Reactive Cysteines. ACS Chem. Biol. 2013, 8, 283–296.
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724.
- De Cesco, S.; Kurian, J.; Dufresne, C.; Mittermaier, A.K.; Moitessier, N. Covalent inhibitors design and discovery. Eur. J. Med. Chem. 2017, 138, 96–114.
- Jackson, P.A.; Widen, J.C.; Harki, D.A.; Brummond, K.M. Covalent Modifiers: A Chemical Perspective on the Reactivity of α, β-Unsaturated Carbonyls with Thiols via Hetero-Michael Addition Reactions. J. Med. Chem. 2017, 60, 839–885.
- Ábrányi-Balogh, P.; Petri, L.; Imre, T.; Szijj, P.; Scarpino, A.; Hrast, M.; Mitrović, A.; Fonovič, U.P.; Németh, K.; Barreteau, H.; et al. A road map for prioritizing warheads for cysteine targeting covalent inhibitors. Eur. J. Med. Chem. 2018, 160, 94–107.
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 2015, 20, 1061–1073.
- Leproult, E.; Barluenga, S.; Moras, D.; Wurtz, J.-M.; Winssinger, N. Cysteine Mapping in Conformationally Distinct Kinase Nucleotide Binding Sites: Application to the Design of Selective Covalent Inhibitors. J. Med. Chem. 2011, 54, 1347–1355.
- Visscher, M.; Arkin, M.R.; Dansen, T.B. Covalent targeting of acquired cysteines in cancer. Curr. Opin. Chem. Biol. 2016, 30, 61–67.
- Zhao, Z.; Liu, Q.; Bliven, S.; Xie, L.; Bourne, P.E. Determining Cysteines Available for Covalent Inhibition Across the Human Kinome. J. Med. Chem. 2017, 60, 2879–2889.
- Bandyopadhyay, A.; Gao, J. Targeting biomolecules with reversible covalent chemistry. Curr. Opin. Chem. Biol. 2016, 34, 110–116.
- Tucker, D.L.; Rule, S.A. A critical appraisal of ibrutinib in the treatment of mantle cell lymphoma and chronic lymphocytic leukemia. Ther. Clin. Risk Manag. 2015, 11, 979–990.
- Tan, L.; Wang, J.; Tanizaki, J.; Huang, Z.; Aref, A.R.; Rusan, M.; Zhu, S.-J.; Zhang, Y.; Ercan, D.; Liao, R.G.; et al. Development of covalent inhibitors that can overcome resistance to first-generation FGFR kinase inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, E4869–E4877.
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620.
- Johansson, M.H. Reversible michael additions: Covalent inhibitors and prodrugs. Mini-Rev. Med. Chem. 2012, 12, 1330–1344.