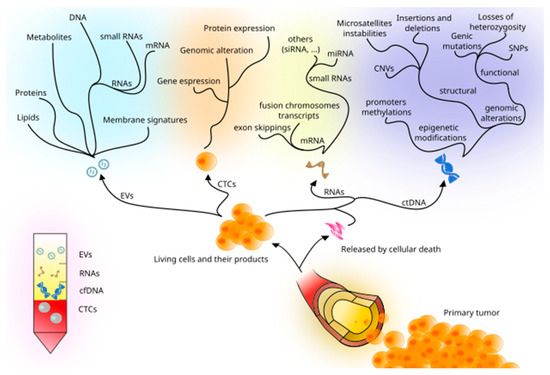
By performing and interpreting an LBx we may get useful information related to the detection of circulating tumor DNA (ctDNA), circulating tumor cells (CTCs), circulating RNAs, and extracellular vesicles (EVs) through a peripheral blood draw.
2. Liquid Biopsy: New Techniques and Biomarkers
Performing an LBx allows physicians and scientists to investigate tumor cells at both the cellular and molecular level: for instance, ctDNA and CTCs are the most studied so far. Recent studies have also included miRNAs, methylation markers, and extracellular vesicles, as emerging and promising alternatives, widening the diagnostic and therapeutic possibilities of LBx (Figure 1).
Figure 1. Principal tumor-derived elements detectable in peripheral blood and the assessable information they may carry.
2.1. cfDNA, ctDNA, and Circulating RNA
Cell-free DNA (cfDNA) consists of total extracellular DNA detectable within the bloodstream, as well as in other biospecimens, such as cerebrospinal and serous fluids [
31,
32,
33].
Cancer cells, as well as non-tumor cells, could shed their genetic material into the bloodstream, as circulating tumor DNA (ctDNA) [
34], which has been proven to be useful in supporting diagnosis, prognosis, disease progression, MRD evaluation, and treatment response, within the context of non-small cell lung cancer (NSCLC) and other solid tumors [
10,
11,
12,
13,
14,
15,
29,
34,
35].
cfDNA is mainly derived from hematopoietic cells circulating in the bloodstream [
36], where its amount is typically limited and poor in terms of quality. It is often necessary to proceed with amplification processes, which may cause artifacts at both PCR and sequencing levels. Only a small fraction of cfDNA is represented by ctDNA, posing a challenge in terms of isolation, processing DNA, and analysis of the generated data. Moreover, discriminating relevant DNA variants from leucocyte hematopoietic clones, such as those found in clonal hematopoiesis of indeterminate potential, may represent another hurdle to overcome [
37,
38], along with the fact that cfDNA could be enriched from other pathological non-tumoral tissues [
39,
40], hence adding another confounding factor in comorbid patients. Furthermore, cfDNA evaluation can be distorted by acute trauma, infections, stroke, exercise, and transplantation [
41,
42,
43,
44,
45], making it necessary to develop better techniques for identifying tumor-specific somatic DNA variants.
In its simplest form, ctDNA analysis can be assimilated into a particular type of genotyping technique, whose goal is to test the presence of a usually restricted number of cancer-related variants. Among the main challenges associated with the ctDNA “genotyping” we may consider: (i) low concentration of circulating DNA; (ii) the existence of fragmented ctDNA; (iii) ctDNA representing only a small fraction of the total cfDNA [
46,
47]. This is particularly true for those samples characterized by a low tumor burden, e.g., at MRD or early diagnosis levels. The latter point is crucial and explains why ctDNA analysis requires dedicated, and often relatively costly techniques, as opposed to conventional genotyping [
48]. Schematically, we can distinguish two scenarios: in the first one, a standard NGS-based approach (e.g., whole-exome or targeted sequencing) is usually adopted using the primary tumor mass, thus identifying the mutational landscape of the tumor. This map can be subsequently used to search, a priori, at nucleotide resolution level, for all the variants that need to be monitored by ctDNA analysis: this is a typical scenario that could apply to the MRD monitoring setting. In the second one, the specific variants that are present in the sample are unknown, which may significantly increase the complexity of the analysis. This typically occurs in early detection screening protocols. In all cases, however, a classical approach based on PCR amplification of the target locus/loci followed by amplicon Sanger sequencing is simply unsuitable for ctDNA analysis, as the sensitivity of Sanger technology for variants occurring at low variant allele frequency (VAF) is too low to be of any practical use in this scenario.
ctDNA analysis typically relies on the following steps: (1) cfDNA extraction; (2) enrichment of the target regions; (3) NGS library preparation; (4) NGS sequencing. cfDNA extraction is usually performed using either spin column- or magnetic bead-based methods [
49,
50,
51,
52]. Despite the spin column-based kits being considered the first choice, when it comes to collecting cfDNA for molecular analyses, they are more expensive and have longer execution times, compared to magnetic bead-based ones [
53].
The enrichment step usually involves the use of targeted gene panels, aiming to investigate disease-related highly recurrent mutated genes. Moreover, in cases when the specific target mutations are known, PCR-based enrichment can be considered as well. In both scenarios, precautions must be taken to avoid preparation artifacts such as amplification-based errors. In terms of the NGS library preparation step, since high sensitivity and specificity are major goals in virtually all ctDNA analyses, several NGS protocols have been recently developed to maximize the reliability of NGS-based ctDNA variant calling [
54,
55,
56]; these include, for instance, CAPPSeq, iDES, and SAFE-seq techniques [
57,
58,
59], whose detailed description goes beyond the purpose of this review. The final NGS sequencing step depends upon the amount of sequencing material, being roughly proportional to the number of profiled genes and the desired depth of the analysis [
60].
In general, one of the significant advantages of NGS techniques is that they are particularly sensitive in spotting single nucleotide variants (SNVs), small insertions and deletions, and copy number variations (CNVs), while the gene fusion and exon skipping detection fall within the competence of PCR and RNA sequencing.
Currently available ctDNA assays use hybrid-capture-based or Amplicon/PCR-amplified-based methodologies and are limited to a strict selection of gene sets [
61]. Ongoing studies are assessing whole genome sequencing as a cheaper, more rapid, and accessible methodology for obtaining significant information from the LBx [
62].
In the context of the transcriptome evaluation, the dysregulation of microRNAs (miRNAs) plays a role in the pathogenesis of several diseases, including cancer, by taking part in post-transcriptional modification of genes related to apoptosis, stress response, mitosis, and cell differentiation [
17,
63,
64,
65,
66]. MiRNAs also circulate in the bloodstream, configuring themselves as promising diagnostic biomarkers and, possibly, therapeutic targets [
67,
68,
69].
Our knowledge about the usefulness of the circulating tumor components and the appropriate techniques to evaluate the messages they carry is still scarce and requires further studies and insightful discoveries to be embedded within the clinical practice setting.
2.2. CTCs
CTCs derive from tumor foci, either primary or metastatic [
70,
71]. They are released from the primary source in form of clusters, but they can also be found as single cells, whose correlation with overall survival and prognosis has been established [
72]. CTCs could provide early information mirroring the primary tumor, such as genomic alterations [
73,
74], and gene and protein expression [
75,
76,
77,
78] characterizing the tumor cells. The existence of sub-clonal CTC populations has been confirmed as related to the cancer metastatic spreading, [
79] leading to possible new ways of diagnosing the disease progression beforehand. CTCs’ rarity, heterogeneity, and the difficulties involved in their analysis make their clinical involvement quite challenging [
80,
81,
82]. Overall, given the ability of CTCs to mirror the tumor of origin, they have been shown to provide useful information about the tumor itself, its metastatic spreading, as well as resistance or sensitivity to therapies [
83,
84,
85].
CTCs detection, unfortunately, requires complex enrichment techniques, on the basis of both biological (i.e., specific antibody affinity) and physical properties (i.e., selecting them through size or deformability) [
86,
87]. Similarly to cfDNA, the amount of CTCs compared to the number of normal nucleated blood cells is very low: this has represented a major challenge that could affect the reliability of the results [
88].
For CTCs detection and counting, FDA has currently approved one platform (CellSearch
® system, Menarini Silicon Biosystems, Firenze, Italy), based on EpCAM
+ CTCs identification [
89].
2.3. Methylation Markers
DNA methylation has been reported to support the pathogenesis and disease progression of several cancer types. For instance, it can enhance the metastatic process, acting on promoter regions related to tumor suppressor genes [
90,
91,
92]. It could also give information about the treatment response [
93]. Since the methylation process could anticipate the manifestation of the full-blown disease [
94], it is reasonable to think to integrate ctDNA methylation sequencing for early diagnosis, in a disease-related targeted way [
95]. Based on this idea, Liu et al. conducted a study with a wide cohort of neoplastic patients, in which this method managed to diagnose more than 50 cancer types across all stages, with great specificity [
96], but with low sensitivity level, particularly in early-stage cases [
97]. The high specificity levels were justified by the rarity of DNA methylation findings in healthy samples’ CTCs and cfDNA [
91,
92,
98,
99]. In parallel, some studies have reported on the use of DNA methylation as a potential novel cancer biomarker, as shown for GSTPI, PITX2, and MGMT, within the context of prostate cancer, lymph node-negative breast cancer, and glioblastomas, respectively [
100].
2.4. Extracellular Vesicles
Cells are capable of releasing vesicles across the extracellular space, as demonstrated within the context of both physiological and pathological conditions. These are heterogeneous particles, delimited by a phospholipid bilayer, responsible for several functions such as cell-to-cell communication [
101], thus supporting the cross-talk between malignant cells and the microenvironment cells [
102], as it has been shown within the specific context of multiple myeloma [
103]. It is possible that, even when CTCs are below the threshold of detection, extracellular vesicles (EVs), for instance, nanovesicles, could be analyzed [
104], and used to support the identification of cancer biomarkers. The content of these nanovesicles, together with lipids, proteins, and metabolites, often consists of small fragments of RNA and DNA [
105]. They can generate from a tumor source, thus reflecting the primary tumor cell-specific mutational status, serving as a diagnostic biomarker [
106,
107]. In contrast to ctDNA, shed through cellular death mechanisms, EV-DNA is actively and selectively released by subclonal tumor cells and is wrapped in a membrane that protects the nucleic acids from a faster degradation, which makes it a more accurate depiction of the tumor and its diversified environment; nevertheless, the technological limitation in isolating EVs and analyzing them singularly prevents, at the moment, this promising opportunity from being adopted within clinical practice [
108].
The dysregulation of microRNAs (miRNAs) plays a role in the pathogenesis of several diseases, including cancer, by taking part in post-transcriptional modification of apoptosis-, stress response-, mitosis-, and cell differentiation-related genes [
17,
63,
64,
65,
66]. miRNAs circulate in the bloodstream both freely and as exosomal miRNAs, configuring themselves as promising diagnostic biomarkers and, possibly, therapeutic targets [
67,
68,
69,
109,
110]. As reported in the literature, exosomal small RNAs (such as miRNAs) represent the main content of EVs [
103,
111,
112].
EVs are usually characterized in terms of both morphology and content; this is achieved using several techniques, such as fluorescence-based platforms, electron microscopy, immunogold labeling, flow cytometry, and mass spectrometry [
113,
114], with great sensitivity and specificity degrees [
103,
115]. In the specific case of exosome characterization, for instance, CD63 and CD81 represent key markers to be investigated at the protein level [
103,
116,
117].