Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biotechnology & Applied Microbiology
Fatty acid oxidation disorders (FAODs) are inborn errors of metabolism (IEMs) caused by defects in the fatty acid (FA) mitochondrial β-oxidation. The most common FAODs are characterized by the accumulation of medium-chain FAs and long-chain (3-hydroxy) FAs (and their carnitine derivatives), respectively.
- inborn errors of metabolism
- FAOD
- MCADD
- LCHADD
- VLCADD
- CPT2D
1. Introduction
Mitochondrial fatty acid (FA) β-oxidation is a complex pathway which occurs in mitochondria to produce energy from lipids. The deficiency in specific transport proteins of FA or enzymes involved in FA β-oxidation is responsible for FA β-oxidation disorders (FAODs) [1]. More than 15 distinct disorders have been described, and the most prevalent FAODs are the medium-chain acyl-CoA dehydrogenase deficiency (MCADD), the long-chain hydroxy acyl-CoA dehydrogenase deficiency (LCHADD), and the very long chain acyl-CoA dehydrogenase deficiency (VLCADD) [2]. The clinical severity and age of onset are highly variable [1]. There is some overlap in the clinical phenotypes of diverse FAODs, such as acute hypoketotic hypoglycaemia and encephalopathy episodes. Although cardiac arrhythmias can occur in both MCADD and long-chain FAODs, cardiomyopathy and myopathy occur only in the latter [3]. Clinical phenotypes, with potential life-threatening manifestations, are the consequence of energy deficiencies and toxic effects of FA and acylcarnitine (CAR) accumulation [4,5]. To manage these diseases, patients need to follow a life-long avoidance of long fasting, a normal/low-fat diet, and carbohydrate and carnitine supplementation [1]. Both the disturbance of FAO and diet modifications can cause changes in the lipidome in FAOD patients. These can have a role in disease pathophysiology, determining clinical manifestations and prognosis.
Lipids have multiple functions in cells/biofluids, namely as an energy source, or as a major components of cell membranes and signalling molecules. Disturbances in the lipid profile have been associated with the pathophysiology of several diseases [6,7,8,9,10]. In FAODs, changes in lipids, namely in free and esterified FAs of plasma, dried blood spots (DBSs), and tissues have been reported. Despite minimal evidence that polar lipids, the main components of cell/organelle membranes and signalling molecules, can be affected in FAODs, the plasticity of the profile of these complex lipids has been scarcely studied [11,12].
2. Mitochondrial Fatty Acid β-Oxidation
The mitochondrial FAO is one of the main pathways for energy production and is important for cell homeostasis [13,14,15,16]. FAO has a major role during metabolic stress, febrile illness, exercise, and fasting conditions, when the glucose levels in the blood decrease, providing an alternative fuel and sparing glucose [13,14,15,16].
The substrates for mitochondrial FAO come from three main sources: (1) FAs from the diet or mobilized from other tissues that enter into the cell via the blood stream, (2) the de novo synthesis of FAs, and (3) FAs released within the cell through the hydrolysis of phospholipids (PLs) and triglycerides (TAGs) [17,18]. A schematic representation of mitochondrial FAO is shown in Figure 1. FAs from the diet and adipose tissue are transported through the blood stream bound to albumin and lipoproteins [19,20]. Upon reaching the target cells, the short- and medium-chain FAs cross the plasmatic membrane by passive diffusion [15]. In contrast, long-chain FAs are actively transported across the cell membrane by specific protein carriers present in the plasmatic membrane, namely FA transport proteins (FATPs), FA translocase (FAT), and FA-binding proteins (FABPs) (Figure 1) [13,15,19]. Once in the cytosol, long-chain FAs are converted into acyl-CoA esters, while short- and medium-chain FAs cross the mitochondrial membrane by passive diffusion and are converted to acyl-CoA esters inside the mitochondrial matrix [15,20,21]. As the mitochondrial membrane is not permeable to long acyl-CoA esters, the carnitine shuttle is required [13,19,22]. It needs the action of three distinct membrane-bound proteins: the carnitine palmitoyl transferase 1 (CPT1), which catalyses the formation of acylcarnitines (CARs) from acyl-CoA esters and free l-carnitine; the mitochondrial carnitine–acylcarnitine translocase (CACT), responsible for the exchange of a CAR for a free l-carnitine molecule from the mitochondrial matrix; and the carnitine palmitoyl transferase 2 (CPT2), which re-esterifies the CAR to the respective acyl-CoA esters [13,15,16,19,23]. Inside the mitochondrial matrix, the activated form of the FA (fatty acyl-CoA) is degraded by β-oxidation into acetyl-CoA units through a cyclic process.
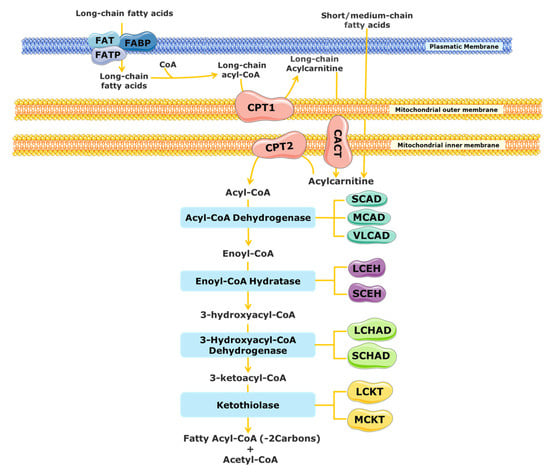
Figure 1. Schematic representation of mitochondrial fatty acid β-oxidation. CACT, carnitine acylcarnitine translocase; CPT, carnitine palmitoyltransferase; FABS, fatty-acid-binding protein; FAT, fatty acid translocase; FATP, fatty acid transport protein; LCHAD, long-chain 3-hydroxyacyl-CoA dehydrogenase; MCAD, medium-chain acyl-CoA dehydrogenase; SCAD, short-chain acyl-CoA dehydrogenase; SCHAD, short-chain 3-hydroxyacyl-CoA dehydrogenase; VLCAD, very long chain acyl-CoA dehydrogenases; LCEH, long-chain enoyl-CoA hydratase; SCEH, short-chain enoyl-CoA hydratase; LCKT, long-chain 3-ketoacyl-CoA thiolase; MCKT, medium-chain ketoacyl-CoA thiolase.
Each β-oxidation cycle involves four enzymatic steps, which results in the release of two-carbon units (as acetyl-CoA) from the FA, with formation of a shorter FA [13]. These steps are catalysed by several enzymes with chain-length specificities, which overlap [15,21]. The first step, the dehydrogenation of the acyl-CoA ester to a trans-2-enoyl-CoA, is catalysed by different acyl-CoA dehydrogenases [15,24]. The short-chain acyl-CoA dehydrogenase (SCAD) acts on substrates with a chain length between C4 and C6, the medium-chain acyl-CoA dehydrogenase (MCAD) acts on C6 to C12 substrates, and the very long chain acyl-CoA dehydrogenase (VLCAD) is active with substrates ranging from C14 to C24 [16,23,25]. Following the initial dehydrogenation, a hydrogenation step of trans-2-enoyl-CoA is catalysed by 2-enoyl-CoA hydratases, producing an l-3-hydroxyacyl-CoA. At least two enzymes can carry out this reaction, the long-chain enoyl-CoA hydratase (LCEH) and the short-chain enoyl-CoA hydratase (SCEH), which are responsible for the hydration of long- and medium/short-chain enoyl-CoA species, respectively [14,23,25,26]. In the third step of the β-oxidation cycle, the resulting l-3-hydroxyacyl-CoA is dehydrogenated to 3-keto-acyl-CoA by a 3-hydroxyacyl-CoA dehydrogenase. The medium/short-chain 3-hydroxyacyl-CoA dehydrogenase (M/SCHAD) is responsible for the reduction of the medium- and short-chain l-3-hydroxyacyl-CoA. The long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD), located at the α-subunit of the mitochondrial trifunctional protein (MTP), is responsible for the reduction of long-chain substrates [14,25,26]. The final step of the FAO cycle is represented by a thiolytic cleavage of the l-3-hydroxyacyl-CoA to yield an acetyl-CoA (or a propionyl-CoA, in the case of 2-methyl-branched-chain FA) and a two-carbon short-chained fatty acyl-CoA. This step is catalysed by the medium-chain 3-ketoacyl-CoA thiolase (MCKT), which acts on substrates between C4 and C12. The long-chain 3-ketoacyl-CoA thiolase (LCKT) is long-chain-specific [14,25,26]. Similar to the LCHAD enzyme, LCEH and LCKT are also part of the MTP [2].
Overall, there are at least twenty-five enzymes and specific transport proteins involved in mitochondrial FAO. Defects in many of them are associated with human diseases, notably FAODs, as will be detailed in the next section.
3. Mitochondrial Fatty Acid β-Oxidation Disorders (FAODs)
FAODs are autosomal recessive genetic defects and represent an important group of inborn errors of metabolism (IEMs). Although individually rare, they are relatively common as a group [16,27]. The most common FAODs can be divided in two groups, depending on the length of the FA affected: medium- and long-chain FAODs [26,28]. The former is MCADD and it is the most prevalent FAOD. The latter group includes several disorders of long-chain dehydrogenases and the FA mitochondrial carnitine transport system. LCHADD and VLCADD are the most prevalent of all long-chain FAODs [27,29]. FAODs have distinct clinical presentations and therapeutic needs [27]. Symptoms are triggered by long fasting, exercise, fever, and other causes of metabolic stress, such as surgery or trauma. Clinical presentation and severity are highly variable and may include acute hypoketotic hypoglycemia, peripherical neuropathy, cardiomyopathy, arrhythmias, hepatopathy, and/or myopathy [3,30].
FAODs are characterized by the accumulation of CARs, namely hexanoylcarnitine (CAR 6:0), octanoylcarnitine (CAR 8:0), decanoylcarnitine (CAR 10:0), and decenoylcarnitine (CAR 10:1 n-6, in MCADD [16,31]; 3-hydroxylcarnitines, namely 3-hydroxyhexadecanoylcarnitine (CAR 16:0;O), 3-hydroxyhexadecenoylcarnitine (CAR 16:1;O), 3-hydroxyoctadecanoylcarnitine (CAR 18:0;O), and 3-hydroxyoctadecenoylcarnitine (CAR 18:1;O) in LCHADD [16,27]; and tetradecenoylcarnitine (CAR 14:1) (the primary marker of this condition), tetradecanoylcarnitine (CAR 14:0), tetradecadienylcarnitine (CAR 14:2), dodecenoylcarnitine (CAR 12:1), and hexadecanoylcarnitine (CAR 16:0) in VLCADD [16,32]. The accumulation of CARs, as well as the respective free FAs, causes lipotoxicity and alters cell homeostasis [4,27].
FAODs can have life-threatening manifestations, namely hypoketotic hypoglycemia, encephalopathy, cardiac arrythmias and cardiomyopathy with heart failure, and rhabdomyolysis crisis with acute renal failure. Early diagnosis and treatment can be effective to prevent most of those complications, reducing morbidity and mortality. Therefore, FAODs are included in the newborn screening (NBS) program of several European countries [7,33]. The NBS of FAODs is based on the quantification of specific CARs in dried blood spots (DBSs) using highly sensitive techniques based on tandem mass spectrometry (MS/MS) for targeted analysis [2,34,35]. In addition to the identification of specific CAR profiles, CAR ratios are also analysed. For example, in VLCADD, the use of the CAR 14:1/CAR 2:0 and CAR 14:1/CAR 12:1 ratio helps to reduce the risk of false-negative results and improve the sensitivity of NBS diagnoses [4,15,36]. Afterwards, a definitive test by enzymatic and/or genetic analysis is performed to confirm the diagnosis [37]. In Table 1, the incidence, clinical presentation, and the characteristic CAR profile used in NBS for some FAODs are presented.
In general, the therapeutic management of FAODs aims to balance the energy deprivation and accumulation of toxic intermediates resultant from these metabolic defects [37]. MCADD patients can usually be managed with avoiding long fasting and with carbohydrate and carnitine supplementation, as needed [38,39]. In long-chain FAODs, a lipid-restricted diet is needed, with an important part of the usual dietary lipids (long-chain) substituted by medium-chain triglycerides containing C8 FAs (MCT-C8). The nutritional guidelines for FAOD therapy differ, depending on the FAOD subtype and the symptomatic/asymptomatic status of the patient [29]. Other therapeutic approaches, such as the use of anaplerotic therapy (triheptanoin) and bezafibrates, can be used in selected cases. Triheptanoin is a triglyceride composed by three odd-chain C7 FAs (MCT-C7) and can be used also as an alternative to MCT-C8 supplementation [31,36,37]. The advantage of using MCT-C7 instead of MCT-C8 is characterized by an anaplerotic effect by propionyl-CoA which can be fed into the citric acid cycle and produce succinyl-CoA, succinate, fumarate, malate, and oxaloacetate, which allows gluconeogenesis [31,36,37]. The bezafibrates represent a group of peroxisome proliferator-activated receptor (PPAR) agonists that are able to reduce lipid levels in human blood [36]. However, more clinical studies are needed to evaluate the effectiveness of these alternative therapeutics. Meanwhile, the dietary approach is the cornerstone for lifelong treatment in many FAODs.
As mentioned above, the inefficient use of FAs as an energy source and the accumulation of specific CARs and FAs in FAODs, as well as the restriction of essential FAs and other lipids in the diet and the MCT supplementation, can alter lipid homeostasis. Indeed, alterations in the lipid diet have been related to changes in the FA profile and membrane lipids, impacting the lipid metabolism and cellular function [40,41]. Moreover, the accumulation of specific CARs and FAs can reduce the FA intake by the cell, increase oxidative stress, and cause oxidative damage to lipids, changing the cellular lipid profile [4]. These changes may be involved in pathophysiology FAODs, as will be detailed in the next section.
Table 1. Some of the defects of the mitochondrial fatty acid β-oxidation with worldwide and Portuguese prevalence, clinical presentations, and newborn screening (NBS) acylcarnitine profile. Adapted from Sim et al. [15], Knottnerus et al. [2], Merritt et al. [31], and El-Gharbaway et al. [42].
| Deficiency | Disorder Abbreviation | Worldwide Prevalence (Portugal in 2020) |
Hypoketotic Hypoglycemia | Rhabdomyolysis | Cardiomyopathy | Skeletal Myopathy | Liver Dysfunction | Encephalopathy | Peripherical Neuropathy | Retinopathy | Acylcarnitine Marker (NBS) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mitochondrial β-oxidation spiral | |||||||||||
| Medium-chain acyl-CoA dehydrogenase | MCADD | 1:4000 to 1:15,000 (1:7005) |
X | - | - | - | X | X | - | - | CAR8:0, CAR8:0/CAR10:0 and CAR8:0/CAR2:0 |
| Very long chain acyl-CoA dehydrogenase | VLCADD | 1:30,000 to 1:100,000 (1:129,272) |
X | X | X | X | X | - | - | - | CAR 14:1, CAR 14:2, CAR 14:1/CAR 2:0 and CAR 14:1/CAR 12:1 |
| Long-chain 3-hydroxyacyl-CoA dehydrogenase | LCHADD | 1:110,000 to 1:150,000 (1:94,800) * |
X | X | X | X | X | - | X | X | CAR 16:0;O, CAR 18:1;O and CAR 18:0;O |
| Carnitine shuttle | |||||||||||
| Carnitine palmitoyl transferase deficiency type 1 | CPT1D | 1:500,000 (1:473,998) |
X | - | - | - | X | - | - | - | Free carnitine/(CAR16:0 + CAR18:0) |
| Carnitine palmitoyl transferase deficiency type 2 | CPT2D | Rare (1:284,399) |
X | X | X | X | X | - | - | - | (CAR16:0 + CAR18:0)/CAR2:0 |
| Carnitine–acylcarnitine translocase | CACTD | Rare (1:284,399) |
X | X | X | X | - | - | - | (CAR16:0 + CAR18:0)/CAR2:0 | |
* This prevalence is referred to general MTP deficiencies (not differentiated based on the CAR profile in the NBS). Isolated LCHAD deficiency is the most common disorder of this complex [42].
This entry is adapted from the peer-reviewed paper 10.3390/ijms232213933
This entry is offline, you can click here to edit this entry!