Several models of prostate cancer (PCa) have been developed, each with their unique applications, advantages, and disadvantages. The shortage of clinically relevant, in vivo models is particularly a large barrier to comprehending the tumor progression observed in PCa. Patient-derived xenografts (PDXs) are models that are derived from biopsy specimens and metastatic lesions from human patients, and allow researchers to understand in vivo physiology as well as tumor heterogeneity. Despite the clinical utility of PDXs, they are also met with limited availability, higher cost, and advanced technical expertise required for use. Organoids, or "mini organs", are clusters of cells grown in vitro that self-organize and differentiate into functional cell types. Organoids referred to as patient-derived organoids (PDOs) can be derived from primary tissue materials, and have also been demonstrated to be derived from PDXs themselves. Due to their lower cost and ease of use, they work well for molecular and mechanistic studies, while still maintaining appropriate tumor heterogeneity and disease modeling.
- prostate cancer
- knockout mouse models
- genetically-engineered mouse models
- xenografts
- PDXs
- organoids
1. Introduction
Historical Timeline of PCa Models
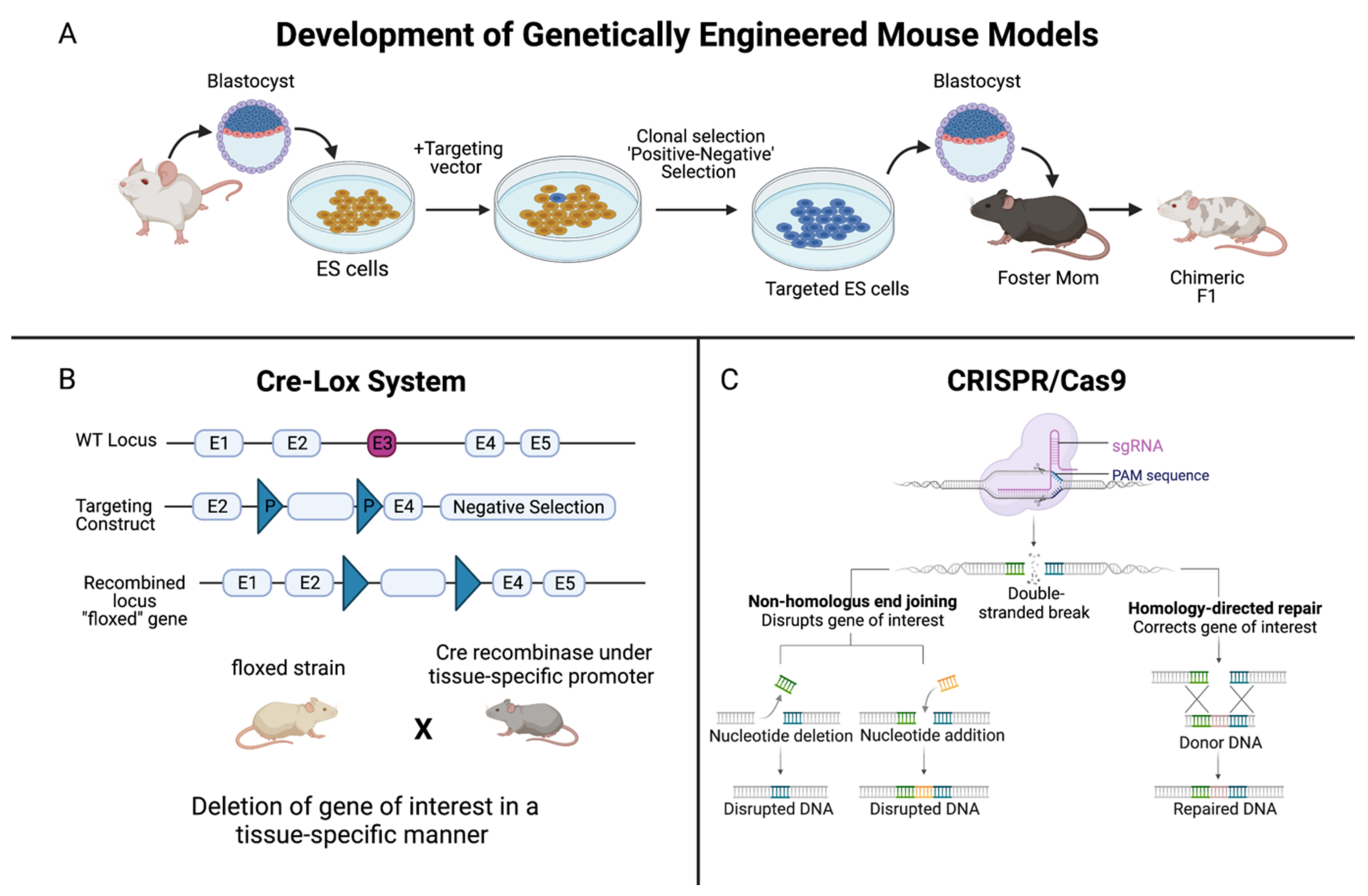
2. Patient-Derived Xenografts and Organoids of PCa
This entry is adapted from the peer-reviewed paper 10.3390/cancers14215321
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33.
- Sanford, K.K.; Hobbs, G.L.; Earle, W.R. The tumor-producing capacity of strain L mouse cells after 10 years in vitro. Cancer Res. 1956, 16, 162–166.
- Scherer, W.F.; Syverton, J.T.; Gey, G.O. Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in a stable strain of human malignant epithelial cells (strain HeLa) derived from an epidermoid carcinoma of the cervix. J. Exp. Med. 1953, 97, 695–710.
- Jedrzejczak-Silicka, M. History of Cell Culture. In New Insights into Cell Culture Technology; Gowder, S.J.T., Ed.; IntechOpen: London, UK, 2017.
- Horoszewicz, J.S.; Leong, S.S.; Kawinski, E.; Karr, J.P.; Rosenthal, H.; Chu, T.M.; Mirand, E.A.; Murphy, G.P. LNCaP model of human prostatic carcinoma. Cancer Res. 1983, 43, 1809–1818.
- Stone, K.R.; Mickey, D.D.; Wunderli, H.; Mickey, G.H.; Paulson, D.F. Isolation of a human prostate carcinoma cell line (DU 145). Int. J. Cancer. 1978, 21, 274–281.
- Kaighn, M.E.; Narayan, K.S.; Ohnuki, Y.; Lechner, J.F.; Jones, L.W. Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Investig. Urol. 1979, 17, 16–23.
- Press Release. Nobel Prize Outreach AB. 2022. Available online: https://www.nobelprize.org/prizes/medicine/2007/press-release/ (accessed on 25 October 2022).
- Nagy, A. Cre recombinase: The universal reagent for genome tailoring. Genesis 2000, 26, 99–109.
- Dow, L.E.; Fisher, J.; O’Rourke, K.P.; Muley, A.; Kastenhuber, E.R.; Livshits, G.; Tschaharganeh, D.F.; Socci, N.D.; Lowe, S.W. Inducible in vivo genome editing with CRISPR-Cas9. Nat. Biotechnol. 2015, 33, 390–394.
- Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; An, P.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562.
- Woo, X.Y.; Giordano, J.; Srivastava, A.; Zhao, Z.M.; Lloyd, M.W.; de Bruijn, R.; Suh, Y.S.; Patidar, R.; Chen, L.; Scherer, S.; et al. Conservation of copy number profiles during engraftment and passaging of patient-derived cancer xenografts. Nat. Genet. 2021, 53, 86–99.
- Nguyen, H.M.; Vessella, R.L.; Morrissey, C.; Brown, L.G.; Coleman, I.M.; Higano, C.S.; Mostaghel, E.A.; Zhang, X.; True, L.D.; Lam, H.M.; et al. LuCaP Prostate Cancer Patient-Derived Xenografts Reflect the Molecular Heterogeneity of Advanced Disease and Serve as Models for Evaluating Cancer Therapeutics. Prostate 2017, 77, 654–671.
- Hao, J.; Ci, X.; Xue, H.; Wu, R.; Dong, X.; Choi, S.Y.C.; He, H.; Wang, Y.; Zhang, F.; Qu, S.; et al. Patient-derived Hormone-naive Prostate Cancer Xenograft Models Reveal Growth Factor Receptor Bound Protein 10 as an Androgen Receptor-repressed Gene Driving the Development of Castration-resistant Prostate Cancer. Eur. Urol. 2018, 73, 949–960.
- Lin, D.; Xue, H.; Wang, Y.; Wu, R.; Watahiki, A.; Dong, X.; Cheng, H.; Wyatt, A.W.; Collins, C.C.; Gout, P.W.; et al. Next generation patient-derived prostate cancer xenograft models. Asian J. Androl. 2014, 16, 407–412.
- Corro, C.; Novellasdemunt, L.; Li, V.S.W. A brief history of organoids. Am. J. Physiol. Cell Physiol. 2020, 319, C151–C165.
- Beshiri, M.L.; Tice, C.M.; Tran, C.; Nguyen, H.M.; Sowalsky, A.G.; Agarwal, S.; Jansson, K.H.; Yang, Q.; McGowen, K.M.; Yin, J.; et al. A PDX/Organoid Biobank of Advanced Prostate Cancers Captures Genomic and Phenotypic Heterogeneity for Disease Modeling and Therapeutic Screening. Clin. Cancer Res. 2018, 24, 4332–4345.
- Karthaus, W.R.; Iaquinta, P.J.; Drost, J.; Gracanin, A.; van Boxtel, R.; Wongvipat, J.; Dowling, C.M.; Gao, D.; Begthel, H.; Sachs, N.; et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 2014, 159, 163–175.
- Puca, L.; Bareja, R.; Prandi, D.; Shaw, R.; Benelli, M.; Karthaus, W.R.; Hess, J.; Sigouros, M.; Donoghue, A.; Kossai, M.; et al. Patient derived organoids to model rare prostate cancer phenotypes. Nat. Commun. 2018, 9, 2404.
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187.