Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Similar to previous pandemics, COVID-19 has been succeeded by well-documented post-infectious sequelae, including chronic fatigue, cough, shortness of breath, myalgia, and concentration difficulties. Dysfunctional efferocytosis has been associated with biological barrier disruption, inflammatory bowel disease (IBD), and a constellation of symptoms reminiscent of long COVID and other fatiguing illnesses.
- cortisol
- HMGB1
- microbial translocation
- SARS-CoV-2
1. Introduction
In the post-pandemic era, residual or long COVID-19 sequelae have been gradually emerging as many patients experience prolonged fatigue, cough, shortness of breath, myalgia, and problems with concentration long after the acute illness phase [1]. From a biological pathway perspective, as both SARS-CoV-2 infection and the associated psychological stress upregulate cortisol, the function of macrophages and natural killer (NK) cells may be impaired, disrupting the clearance of senescent, damaged, or virus-infected cells. This may lead to biological barrier dysfunction and chronic fatigue, phenomena well-documented in cancer survivors treated with cellular senescence-inducing chemotherapy or radiation [2,3,4,5].
At the molecular level, upregulated cortisol lowers the expression of claudin-1 (CLDN1), an intestinal tight junction protein, facilitating microbial translocation outside of the gastrointestinal (GI) tract [6]. In the central nervous system (CNS), P-glycoprotein (P-gp), a cortisol substrate, functions as a gatekeeper of the blood–brain barrier (BBB), likely accounting for hypercortisolemia-increased barrier permeability [7,8,9]. Interestingly, in the GI tract, gut microbiota and lipopolysaccharide (LPS), a cell wall component of Gram-negative bacteria, regulate P-gp, connecting this protein to microbial translocation [10,11].
Recent studies have found a direct relationship between circulating levels of cortisol and premature cellular senescence, a phenotype characterized by permanent cell cycle arrest, active metabolism, and a detrimental secretome [12]. The accumulation of senescent cells was found to be associated not only with organismal aging but also with chronic fatigue, pain, and depression, documented in cancer survivors [2]. On the other hand, enhanced elimination of senescent cells via senotherapeutics can correct barrier dysfunction, lowering fatigue [13,14,15].
NK cells and macrophages can execute the phagocytic engulfment (efferocytosis) of damaged or dead cells, including malignant, virus-infected, and senescent cells. Dysfunctional efferocytosis has been associated with biological barrier disruption, inflammatory bowel disease (IBD), and a constellation of symptoms reminiscent of long COVID and other fatiguing illnesses [16,17,18,19,20,21]. Indeed, NK cell dysfunction is one of the most consistent findings in long COVID-19, myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS), Gulf War illnesses (GWI), and fibromyalgia (FM), linking these pathologies to dysfunctional efferocytosis [22,23,24,25,26,27]. Moreover, NK cells had been previously linked to fatigue-associated thyroid, adrenal, hypothalamic, and pituitary disorders, thus connecting these neuroendocrine -related pathologies with dysfunctional immunity [28,29,30,31,32]. Furthermore, NK cells express estrogen, prolactin, and cortisol receptors as well as a functional renin-angiotensin-system (RAS), rendering them sensitive to hormonal fluctuations [33,34,35]. NK cells are capable of paracrine signaling and they secrete biomolecules, including perforin, granzyme B, and the high mobility group box 1 protein (HMGB1) that can facilitate the elimination of damaged, senescent, and/or malignant cells [36,37,38].
Microbial translocation markers, such as LPS, lipopolysaccharide binding protein (LPB), soluble CD14 (sCD14), and HMGB1, were found to be elevated in long COVID, highlighting the role of dysfunctional gut barrier in this condition [30,31,32,33,34,35,36,37,38,39,40,41]. Indeed, accumulation of senescent cells has been associated with HMGB1 spillover into the extracellular space where it can act as an inflammagen and barrier disruptor [42,43,44,45,46]. Therefore, upregulated HMGB1, documented in ME/CFS, FM, and GWI, and COVID-19, directly links dysfunctional efferocytosis to chronic fatigue [47,48,49,50]. For example, gut HMGB1 disrupts the barrier tight junctions and is considered a biomarker of inflammatory bowel disease (IBD), a condition associated with both fatigue and increased GI tract permeability. This may explain certain SARS-CoV-2 bacteriophage-like properties as this virus can likely penetrate the microbial cell walls [51,52,53,54,55,56,57].
2. Efferocytosis and Biological Barriers
Each day billions of cells throughout the body undergo apoptosis and are removed by professional phagocytes, macrophages, monocytes, and neutrophils as well as non-professional phagocytes, including the intestinal epithelial cells (IECs) and the M2 microglia in the BBB [58,59,60]. Professional and non-professional phagocytes are assisted by NK cells that can eliminate defective and pathogen-infected cells without prior sensitization [61,62,63]. NK cells maintain the integrity of BBB and the intestinal barrier as they can promptly clear damaged cells, preventing inflammation and barrier disruption [64,65]. To accomplish this, NK cells perforate the membrane of targeted cells by releasing HMGB1, perforin, and granzyme, triggering apoptosis by Ca2+ influx [66,67]. Upregulated cytosolic Ca2+ is known to activate TMEM16F, an enzyme that flips phosphatidylserine (PS) to the outer leaflet of the cell membrane, providing a distress signal, that attracts immune cells, promoting phagocytosis [68]. Exposed PS (ePS) comprises an “eat me” or “fuse with me” signal that can lead to either cell death or syncytia formation, depending on the degree of cell membrane damage [69]. For example, less damaged cells can fuse with each other for protection, a phenomenon documented in in many tissues, including the CNS [70] (Figure 1). Several studies have demonstrated that senescent and cancer cells can avoid elimination by expressing CD47, a “don’t eat me” signal, that inhibits phagocytosis [71,72,73].
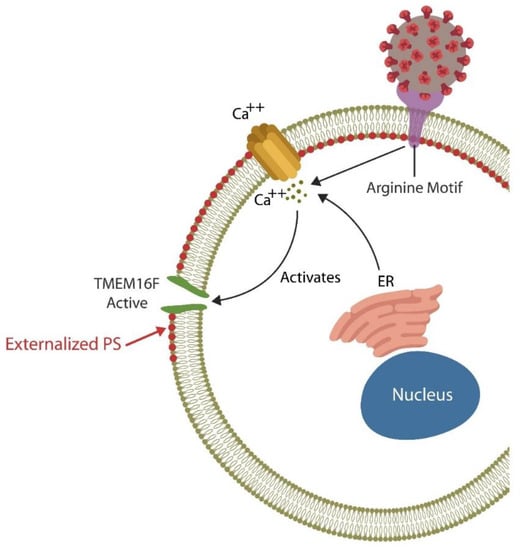
Figure 1. The SARS-CoV-2 receptor binding site (RBS) contains a double arginine insert (PRRA) or arginine motif, that perforates the cell membrane, triggering Ca2+ influx from both the endoplasmic reticulum (ER) and the extracellular compartment. Upregulated cytosolic Ca2+ activates TMEN16F, externalizing phosphatidylserine (ePS), an “eat me” or “fuse me” signal that leads to cell death (if the damage is irreparable) or cell–cell fusion (if the cell can be repaired). Cell–cell fusion or syncytia formation induces premature cellular senescence, disrupting biological barriers. The virus benefits from ePS as this comprises a global immunosuppressive signal, allowing its undetected entry into host cells.
2.1. Blood–Brain Barrier
The BBB, a highly regulated interface between the circulatory system and the CNS, consists of cerebral endothelial cells (ECs) that regulate the inward and outward movement of molecules and ions into the CNS [74]. BBB disruption enables viral entry into the brain along with inflammatory cells, and/or deleterious molecules that can trigger infection. Indeed, members of at least 11 viral families, including the human immunodeficiency virus-1 (HIV-1), T-cell leukemia virus, lymphocytic choriomeningitis virus, West Nile virus, and others, can enter the brain, causing encephalitis [75,76,77].
COVID-19-mediated accumulation of senescent ECs compromises the BBB, allowing viral and microbial access to the CNS [78]. In contrast, enhanced elimination of senescent cells via senolytic drugs decreases COVID-19 mortality in rodents, highlighting the role of senescent cells in BBB dysfunction [79,80]. In addition, the S protein of the SARS-CoV-2 virus was demonstrated to directly bind bacterial LPS, outlining a virus-mediated mechanism of endotoxin entry into the CNS [40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81]. LPS-induced neuroinflammation has been associated with microglial fusion and multinucleation, generating highly phagocytic phenotypes that can cause collateral damage by eliminating viable neurons [82,83]. Indeed, long COVID has been associated with LPS-activated microglia (M1 phenotype), neuroinflammation and neuronal death [82,83,84,85]. On the other hand, the M2 microglial phenotype has been shown to repair the damage, protecting the neurons [58].
2.2. Intestinal Barrier
Many viruses, including SARS-CoV-2, enhance infectivity by usurping both the physiological cell–cell fusion and efferocytosis, disrupting biological barriers [75,86]. For example, the SARS-CoV-2 virus thrives in infected cells and likely inhibits their clearance, causing inflammation and barrier dysfunction [87]. In addition, SARS-CoV-2 promotes pathological cell–cell fusion and syncytia formation by generating cell membrane pores via the PRRA (proline-arginine-arginine-alanine) motif situated at the furin-cleavage site (FCS). This system is reminiscent of microbial twin arginine translocation pathway, a pore-forming mechanism implicated in bacterial virulence [88,89,90]. Moreover, SARS-CoV-2 fusion with Mycoplasma, an arginine dependent microorganism, may explain the high comorbidity of these very different infections [91]. Cell membrane pores lead to ePS, a global immunosuppressive signal, that helps the virus exploit host defenses [92]. The subsequent, syncytia formation can then induce premature cellular senescence and the release of senescence-associated secretory phenotype (SASP), a pathological secretome that disrupts endothelial barriers by promoting premature ECs senescence, a phenotype documented in both ME/CFS and COVID-19 [42,93,94,95].
Senescence-induced pathological syncytia can trigger lymphopenia by cell-in-cell phenomena, elimination of viable lymphocytes, including NK cells, a frequent finding in ME/CFS [96,97,98]. In addition, as cellular senescence upregulates HMGB1, it may further predispose to fatiguing disorders [44,47,48,49,99,100,101,102].
In the GI tract, IECs comprise a single layer of tightly linked columnar cells that are short-lived and need to be replaced every 4 to 5 days to maintain an adequate barrier function [103]. Moreover, IECs acting as non-professional phagocytes, can engulf the translocating microbes and/or antigens, preventing microbial translocation outside the GI tract [104]. However, accumulation of uncleared, defective IECs can trigger inflammation, predisposing to IBD and other illnesses marked by dysfunctional barrier [105]. Macrophages, intestinal NK cells, and Paneth cells contribute to barrier integrity by promptly removing damaged IECs, thus averting necrosis and inflammation-mediated pathology [21,106,107,108]. Interestingly, IECs were demonstrated to produce cortisol, a steroid hormone that lowers the expression of CLDN1 that in return increases intestinal permeability [109,110,111].
The role of ANG II: COVID-19-upregulated angiotensin II (ANG II) can disrupt efferocytosis inducing ECs senescence and vascular barrier dysfunction via angiotensin II type 1 receptors (AT-1Rs) which can enhance both cortisol and HMGB1 production, [112,113] (Figure 2).
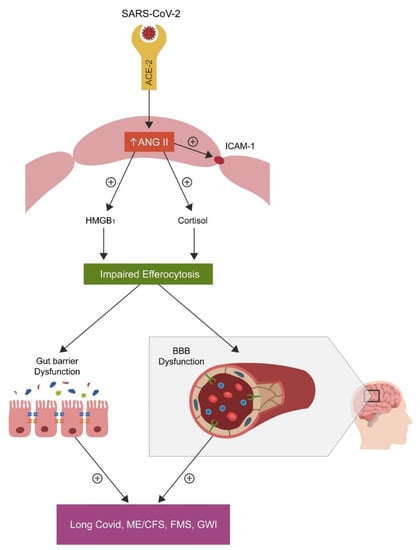
Figure 2. SARS-CoV-2 attachment to ACE-2, blocks this enzyme, causing angiotensin II (ANG II) accumulation by inhibiting its hydrolysis. Upregulated ANG II, increases both cortisol and HMGB1, disrupting the efferocytosis of senescent cells. Accumulation of senescent cells triggers inflammation and biological barrier disruption, a common pathology found not only in the disorders marked by chronic fatigue but also in neuropsychiatric and autoimmune diseases.
Increased cytosolic ANG II disrupts the function of NK cells and, as these cells express a viable RAS, including ACE-2, the virus may directly infect these immune cells [114,115]. In the CNS, ANG II disrupts the BBB via AT1 receptors; in contrast, Losartan, an AT1 antagonist, can repair this and the intestinal biological barrier [116,117].
This entry is adapted from the peer-reviewed paper 10.3390/endocrines3040058
This entry is offline, you can click here to edit this entry!