Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Infectious Diseases
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) induces immune-mediated type 1 interferon (IFN-1) production, the pathophysiology of which involves sterile alpha motif and histidine-aspartate domain-containing protein 1 (SAMHD1) tetramerization and the cytosolic DNA sensor cyclic-GMP-AMP synthase (cGAS)–stimulator of interferon genes (STING) signaling pathway.
- ACE2 (angiotensin-converting enzyme 2)
- aspirin
- cGAS–STING (cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) synthase (cGAS)–stimu
1. SAMHD1 Tetramerization Yields the Catalytically Active Tetramer: SARS-CoV-2 Might Use CDK2 to Phosphorylate SAMHD1
The sterile alpha motif (SAM) is a protein interaction domain involved in developmental regulation. SAM is an evolutionarily conserved protein-binding domain that regulates numerous developmental processes among diverse eukaryotes [13]. SAM and histidine-aspartate domain (HD)-containing protein 1 (SAMHD1) deplete the pool of deoxynucleoside triphosphates (dNTPs) available to a reverse transcriptase for viral cDNA synthesis and thus prevent viral replication [14]. SAMHD1 protects cells from viral infections and operates at stalled replication forks to prevent IFN induction, a significant regulator of dNTP concentrations in human cells [15]. SAMHD1 is a cellular enzyme that drains intracellular dNTPs.
SAMHD1 can restrict retroviruses (human immunodeficiency virus-1, HIV-1). Mutations in SAMHD1 are linked to the pathogenesis of chronic lymphocytic leukemia and Aicardi–Goutières syndrome (AGS). SAMHD1 holds a target motif for cyclin-dependent kinase 1 (CDK1), whose activity is needed for SAMHD1 phosphorylation. SAMHD1 is phosphorylated at residue threonine 592 (T592) in cycling cells, and phosphorylated SAMHD1 on T592 cannot block retroviral infection. Phosphorylation modulates SAMHD1 to block retroviral infection without affecting its ability to decrease cellular dNTP levels [16]. A phosphomimetic mutation surrounding T592 triggered electrostatic repulsion from a distinct negatively charged environment. This resulted in a significant decrease in active SAMHD1 tetramers; hence, the dNTPase activity was substantially decreased. SAMHD1 phosphorylation at residue T592 may modulate its cellular and antiviral functions [17].
Apoenzymes are catalytically inactive, whereas holoenzymes are catalytically active. Inactive apo-SAMHD1 interconverts between monomers and dimers. The binding of deoxyguanosine triphosphates (dGTP) to four allosteric sites promotes tetramerization. It induces a conformational change in the substrate-binding pocket to yield the catalytically active tetramer [18]. The binding sites are plastic, and the allosteric binding sites can adjust oligonucleotides in place of the allosteric activators GTP and dNTP. The binding of G-nucleotide-containing oligonucleotides in the presence of GTP and dNTPs promotes the formation of a specific tetramer with mixed occupancy of the allosteric sites responsible for the antiretroviral activity of SAMHD1 [19].
2. cGAS–STING Signaling
The SAMHD1 tetramer structure could provide a mechanistic understanding of its rapid function in SARS-CoV-2. SAMHD1 negatively regulates the interferon (IFN)-1 signaling pathway: the elevated innate immune response and IFN activation upon genetic loss of SAMHD1 effectively suppress SARS-CoV-2 replication [20]. The spontaneous IFN response requires the cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) synthase (cGAS)–stimulator of interferon genes (STING) cytosolic DNA-sensing pathway in SAMHD1-deficient cells in mice. SAMHD1 mutations cause autoinflammatory AGS, characterized by chronic type I IFN secretion in the absence of infection with exogenous viruses [21] and typified by early onset brain disease [22]. IFN gamma (γ) is a dimerized soluble cytokine, and its signaling is upregulated in the COVID-19 human neurovascular unit [23]. SARS-CoV-2 evokes a response that requires the strong induction of a subclass of cytokines, including type I and, obviously, type III IFN and a few chemokines, such as those produced in response to influenza A virus and, in particular, respiratory syncytial virus [15,24]. SAMHD1 limits the virus-induced production of IFNs and the induction of costimulatory markers during lentivirus infection in myeloid cells. This programmed myeloid cell activation requires reverse transcription, cGAS–STING, and signaling via the interferon receptor. SAMHD1 reduced the stimulation of virus-specific cytotoxic T cells in vivo and limited the induction of antigen-specific T cell responses in vivo. SAMHD1 controls viral infection through innate and adaptive immunity at the level of the infected cell [25].
cGAS catalyzes the production of cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) upon sensing cytosolic DNA, which activates STING–tank-binding kinase 1 (TBK1)–interferon regulatory factor 3 (IRF3) signaling. cGAS is present in the nucleus, and nuclear cGAS is sequestered at chromatin in an inactive state. It recruits protein arginine methyltransferase 5 (Prmt5), facilitating IRF3 access upon viral infection. Nuclear-localized cGAS, in innate immunity, interacts with Prmt5 to catalyze the symmetric dimethylation of histone H3 arginine 2 at IRF3-responsive genes (Ifnb and Ifna4) [26]. Active cGAS produces cyclic GMP-AMP (cGAMP), which binds to STING. STING relocalizes to the perinuclear Golgi and forms a clustered platform where the TBK1 kinase phosphorylates the transcription factor IRF3. Phosphorylated IRF3 enters the nucleus and, along with the nuclear factor kappa-light-chain-enhancer of activated B cells, triggers the expression of type I IFN and proinflammatory cytokine genes [27]. cGAS–STING has been identified as a significant nucleic acid recognition gene. cGAS typically resides as an inactive protein in the cell and is activated upon binding to aberrant DNA. Activated cGAS then synthesizes 2′,3′-cGAMP, which acts as a secondary messenger activating STING. cGAS–STING responds to foreign DNA from viruses and bacteria and to mitochondrial and genomic self-DNA, which enters the cytosol from senescent or dying cells [28]. A SARS-CoV-2 infection could induce syncytia formation within cells expressing ACE2 and the SARS-CoV-2 spike protein, producing micronuclei at an average rate of approximately four per syncytium (>93%). These micronuclei are expressed with a high activation level for the DNA damage response and cGAS–STING signaling. These signaling pathways are associated with cellular catastrophe and aberrant immune activation at the cellular and molecular levels. Activated STING triggers membrane permeabilization and thus lysosomal cell death [29] (Figure 1).
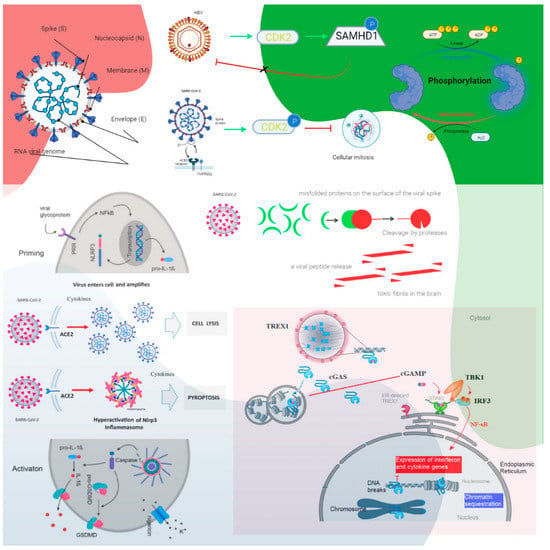
Figure 1. The roles of cyclin-dependent kinase and cGAS–STING signaling. Cyclin-dependent kinase 1 (CDK1), CDK2, and CDK6 inhibit SAMHD1 antiviral function via phosphorylation and inactivation. HBV recruits cyclin E2 to bind CDK2 and further phosphorylates SAMHD1 to abrogate its restriction of HBV replication. SARS-CoV-1 suppresses the activity of cyclin d-CDK4 and cyclin A/E–CDK2 complexes, and SARS-CoV-2 enhances the phosphorylation of CDK2 to inhibit cellular mitosis. Pattern recognition receptors are essential in sensing pathogen-associated molecular patterns (PAMPs) and DAMPs. For example, Toll-like receptors (TLRs) recognize a variety of PAMPs and DAMPs that initiate the inflammatory process via NF-κB (nuclear factor-kappa B cells) and the synthesis and release of cytokines and IFNs. Inflammasomes are a separate class of intracellularly expressed pattern recognition receptors (PRRs) that recognize nucleic acids and mediate proinflammatory responses. Cell surface–expressed ACE2 and TLR4 increase the NLRP3 inflammasome downstream mediator caspase-1, and exposure to spike proteins upregulates the protein expression involved in the positive stimulation of TLR4 signaling and the inflammasome pathway. The induction of neuroinflammation in microglia is mediated through the activation of NF-κB and p38 MAPK (mitogen-activated protein kinase), possibly due to TLR4 activation. TLR4 has been found to play a critical role as a mediator of the neurotoxicity induced by α-synuclein oligomers. Misfolded α-synuclein (α-Syn) induces inflammatory responses, and extracellular α-Syn can activate proinflammatory TLR4 pathways in astrocytes, but α-Syn uptake is independent of TLR4. The interaction between TLR4 and the SARS-CoV-2 spike protein can trigger an intracellular TLR4 signaling cascade. NF-kB’s transcriptional activation of specific genes induces the release of proinflammatory cytokines, which can cause neuronal damage and the pathological modification of α-Syn. Serum neurofilament light chain (NFL) is a biomarker of neuronal injury. NFL was higher in COVID-19 patients than in the control groups. Higher NFL levels were associated with neuronal injury. This is common in critically ill patients. The hyperinflammatory state of COVID-19 has high levels of proinflammatory cytokines, and this might have triggered central nervous system neuroinflammation through the activation of astrocytes and microglia, which could have facilitated prion-like pathology. In addition, similar to other prion proteins, the spike protein also contains several prionogenic domains. Thus, direct toxic action of the spike protein, triggering a neurodegenerative condition mimicking a prion disease-like pathology, is also possible. The biological implications of nuclear cGAS and its interaction with chromatin, including various mechanisms for nuclear cGAS inhibition, the release of chromatin-bound cGAS, the regulation of different cGAS pools in the cell, and chromatin structure/chromatin protein effects on cGAS activation, could lead to cGAS-induced autoimmunity. Cytosolic DNA recognition leads to active cGAS by clustering and forming large liquid—liquid phase-separated cGAS–DNA condensates, excluding the ER-directed exonuclease three-prime repair exonuclease 1 (TREX1). Nuclear cGAS is sequestered at chromatin in an inactive state. Active cGAS produces cyclic GMP-AMP (cGAMP), which binds to STING. STING relocalizes to the perinuclear Golgi and forms a clustered platform where the tank-binding kinase 1 (TBK1) kinase phosphorylates the transcription factor IRF3 (interferon regulatory factor 3). Phosphorylated IRF3 enters the nucleus and, along with NF-κB, triggers the expression of type I interferon and proinflammatory cytokine genes (reproduced from [27]) (HBV, hepatitis B virus; P, phosphate; the green arrows indicate enhancement and the dashed arrow indicates the effect is uncertain; CDK, cyclin-dependent kinase; SAMHD1, sterile alpha motif histidine-aspartic acid domain-containing protein 1).
The activation of cGAS–STING can trigger IRF3-type I IFN and autophagy-mediated antiviral activity. SARS-CoV-2 ORF3a can block viral evasion of the STING-triggered autophagy-mediated antiviral function. It can interact with STING and disrupt the STING-light chain 3 (LC3) interaction but not IRF3-type I IFN induction [30]. SARS-CoV-2 infection also activates cGAS–STING signaling by stimulating micronuclei formation during the process of syncytia, and SARS-CoV-2 open reading frame 10 (ORF10) targets STING to antagonize IFN activation. Its overexpression inhibits cGAS–STING-induced IRF3 phosphorylation, translocation, and subsequent IFN induction. In addition, it interacts with STING, attenuates the STING–TBK1 association, and impairs STING oligomerization and aggregation and STING-mediated autophagy. As a result, it prevents the endoplasmic reticulum (ER)-to-Golgi trafficking of STING by anchoring STING in the ER [31].
Profiling skin manifestations, a STING-dependent type I IFN signature is mediated by macrophages adjacent to areas of endothelial cell damage, and cGAS–STING activity was detected in lungs from COVID-19 patients with prominent tissue destruction, which was associated with IFN-1 responses. A lung-on-chip model with SARS-CoV-2 infection activates cGAS–STING signaling in endothelial cells through mitochondrial DNA release. A STING inhibitor shows anti-inflammatory potential by alleviating detrimental immune responses [32]. Among patients with mild disease in a second phase 2 trial between Peginterferon Lambda-1a and placebo groups, it improved symptoms or other clinical metrics of the odds of achieving viral clearance by day 7 [12].
3. Immunological-Induced Engram Pathway
SARS-CoV-2 may move from the periphery into the CNS through neurons or the vagus nerve from the lungs or gut [33,34]. In addition, SARS-CoV-2 may disrupt the blood–endothelial barrier by damaging the choroid plexus epithelium due to cytokine storms and systemic inflammation through neuronal cell-surface receptors [23,35] (Figure 2).
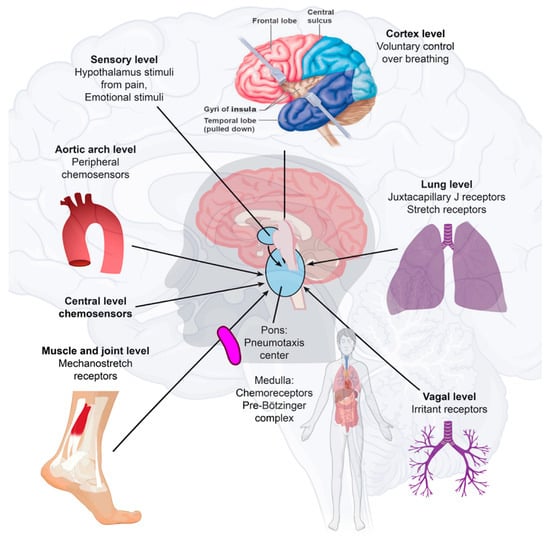
Figure 2. Immune-to-brain communication. The perivascular space contains a single or double layer of invaginated pia in the brain, forming an interstitial fluid-filled space representing an extension of the extracellular fluid space around the intracranial vessels as they move down into the brain parenchyma. Human sensory stimuli affect the breathing sensation passing through the cerebral cortex and hypothalamus. The respiratory muscles are not purposely activated in healthy breathing. Neuropilin-1 (a pleiotropic single-transmembrane coreceptor for class-3 semaphorins and vascular endothelial growth factors) has been confirmed as a coreceptor that facilitates SARS-CoV-2 infection into cells and may be expressed in the brainstem. Activated microglia were observed in the olfactory bulb, midbrain (particularly in the substantia nigra), hindbrain, dorsal motor nucleus of the vagus nerve, and pre-Bötzinger complex in the medulla. Neuroinflammation with microgliosis and T-cell infiltration in COVID-19 brains was significantly greater than that in patients who did not have COVID-19. This might trigger inflammasomes and pyroptosis in the CNS. The pre-Bötzinger complex involvement in the brainstem could account for the respiratory failure and sudden high death rate of COVID-19 ARDS patients. The neurovirulent potential of SARS-CoV-2 is not restricted to patients with mild or severe diseases that can develop neurological complications. Microglia and astrocytes play specific and dynamic roles during immune activation. This immune-to-brain communication occurs when glia, microglia, and astrocytes interpret and propagate inflammatory signals in the brain, and influence physiological and behavioral-change responses. Activating the peripheral immune system with a coordinated brain response elicits and influences physiological and behavioral responses in the immunological memory engram pathway.
In many cases, there is no direct virus attack on vulnerable structures. Instead, the neurological disease manifestations may be due to an immune reaction against the virus, and pre-existing neurological disease may become clinically evident or worsen with COVID-19, which explains why various nervous system manifestations react favorably to immune suppression or immune modulation [38,42]. The insular cortex stores immune-related pieces of information. The chemogenetic reactivation of these neuronal bands was sufficient to broadly retrieve the inflammatory state of these neurons depicted in the insular cortex [43]. These immunological memory engrams, as memory traces, can restore the initial disease state [44]. The brain remembers immune challenges, regulates peripheral immunity, and can exacerbate the process of immune reactions [45]. Microglia and astrocytes play specific and dynamic roles during immune activation. This immune-to-brain communication occurs when glia, microglia, and astrocytes interpret and propagate inflammatory signals in the brain and influence physiological and behavioral-change responses. Astrocytes can develop an immunosenescent profile with age. Astrocyte immunosenescence and deficits in IL-10 signaling in the aged brain disrupt the regulation of microglia following innate immune activation [46,47].
This entry is adapted from the peer-reviewed paper 10.3390/ijms232113260
This entry is offline, you can click here to edit this entry!