Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Acetylation, also known as acetylation or acetylation, refers to the chemical reaction of adding an acetyl functional group to an organic compound. Conversely, the reaction in which the acetyl group is removed is called deacetylation or deacetylation. Acetylation of proteins is a post-translational modification.
- DNA damage response
- post-translational modification
- lipid metabolites
- acetylation
1. Acetylation of Histones and Non-Histone Proteins
In eukaryotic cells, chromatin refers to a linear, complex structure composed of histone, non-histone, DNA, and a small amount of RNA and is located in the nucleus. Histones are alkaline proteins that bind to DNA in eukaryotic cells. They are composed of four core histone families (H2A, H2B, H3, H4) and a linker family (H1). Core histones are mainly responsible for binding to DNA to form nucleosomes, which represent the structural units of chromatin, while linker histones are responsible for binding with nucleosomes to form the next chromatin unit. Thus, histones play an important role in maintaining chromatin structure. Non-histone chromatin refers to other proteins that can bind to chromatin, including chromatin structural proteins, enzymes, and a small number of regulatory proteins. Although non-histones are less common in chromatin than histones, there a wide range of types, and they serve complex functions, such as regulating gene expression and maintaining chromatin structure. There are more basic amino acids in histones than acidic amino acids, but the ratio of basic to acidic amino acids in non-histones varies across situations and contexts.
Histone acetylation is a dynamic and reversible protein PTM and may be the most well-understood. Histone acetyltransferases (HATs, also referred to as lysine acetyltransferases) and histone deacetylases (HDACs, also known as lysine deacetylases) catalyze the addition or removal of acetyl groups, respectively, to lysine residues on both histone and non-histone proteins [29]. Acetylation occurs on amino-terminal proteins and on the O-junctions of serine and threonine, but for the purposes of this review, acetylation refers only to N ε-lysine acetylation (Kac) (defined as the deposition of an acetyl group onto the epsilon amino group of lysine; Figure 1) [30]. It was found that, by stable isotope tracking and acetylation proteomic analysis, more than 90% of acetylation modification on histone lysine is derived from the carbon of fatty acids [26]. Acetyl-CoA in mitochondria from fatty acid β-oxidation is converted to citrate, an intermediate of the tricarboxylic acid cycle. Citrate is exported out of mitochondria by citate transporter and subsequently cleaved by ATP-citrate lyase (ACLY) into acetyl-CoA [31].
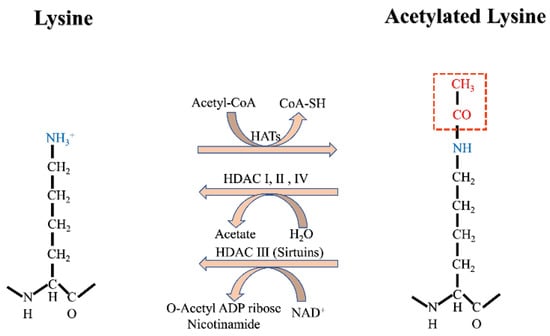
Figure 1. Acetylation modification mechanism.
Acetylation of histones by HATs relaxes the structure of chromatin and increases DNA accessibility, which results in increased gene transcription. The effect of HDACs is the opposite, with deacetylation making the structure of chromatin more compact and inhibiting transcription. The histone code hypothesis suggests that histone modification can be explained by chromatin-binding proteins called “readers,” which distinguish between modified and unmodified nucleosomes and then decide on DNA transcription or other related downstream events [32]. At present, known domains with the ability to recognize or read acetylated lysine residues include YAF9, ENL, AF9, Taf14, and Sas5 YEATS domains, and tandem homologous domains (PHDs, also known as double PHD finger (DPF) domains) in plants [33]. In addition to histones, mass spectrometry has also revealed that acetylation occurs on non-histone proteins, some of which participate in protein folding, protein degradation, and chromatin structure adjustment [34].
The exact number of HATs in the human proteome remains unknown. However, the histone HATs that have been identified thus far include the P160, P300/CBP, TAFII230, MYST, GNAT, and PCAF families, with the current classification system primarily being based on homology with the original enzyme sequence found in yeast. In addition, a total of 13 non-histone HATs have also been identified, and these are roughly divided into three families: GCN5, P300, and MYST. These include histone acetyltransferase 1 (HAT1, also known as KAT1), α-tubulin N-acetyltransferase 1 (TAT1, also known as ATAT1) [35], and establishment of cohesion 1 homology 1 (ESCO1) and ESCO2, which all serve different functions. At present, non-histone HAT specificity is thought to be determined by the accessibility of lysine in their substrate proteins, specific subcellular localization, and interacting proteins. For example, with the exception of TAT1, HATs are primarily located in the cell nuclei. Most HAT substrates do not overlap, but some functionally similar HATs can acetylate the same sites: for example, CREB-binding proteins (CBP, also known as KAT3A) and p300 (also known as KAT3B) both acetylate histone H3Lys18 (H3K18) and H3K27.
HDACs, meanwhile, are divided into four categories based on their sequence similarity and degree of phylogenetic conservation. The class I HDACs include HDAC1, 2, 3, and 8; the class II HDACs include HDAC4, 5, 6, 7, 9, and 10 (being further subdivided into class IIa and IIb); the class III HDACs are known as the sirtuins and include SIRT1-7; while the fourth category currently includes HDAC11 only [36]. With the exception of class III HDACS, which are NAD+-dependent, the other HDAC classes are all zinc-dependent. Zinc-dependent HDACs contains a deacetylase domain which is highly conserved and are often referred to as classical HDACs. While class I and IV HDACs are localized in the nucleus, class IIb HDACs are distributed in the cytoplasm. Class IIa HDACs are mainly distributed in the nucleus but are exported to the cytoplasm when activated. The sirtuins are distributed in different locations in the cell as follows: cytoplasm (SIRT2), mitochondria (SIRT3, SIRT4, and SIRT5), nucleus (SIRT1 and SIRT6), and nucleolus (SIRT7).
It is worth noting that most non-histone deacetylases have limited or no deacetylase activity or primarily perform other types of acylation. For example, SIRT4 [37] removes the acyl group from hydroxymethyl glutaryl lysine, SIRT5 [38] acts as a demalonylase and a deglutarylase, and SIRT6 acts as a long-chain fatty acid deacylase. Class IIa HDACs lack obvious catalytic activity, primarily due to the alterations in the conserved amino acids found in their catalytic pocket. Recent studies have found that lymphoid augmenter factor 1 (LEF1) [39] and T cell-specific transcription factor 1 (TCF1, also known as Tcf7) [40] also exert HDAC activity and that these two transcription factors are involved in the regulation of the WNT signaling pathway [41]. The overall sequences of LEF1 and TCF1 are very different from HDAC8, but their functions are similar.
Recent studies have shown that acetylation can also occur through non-enzymatic mechanisms [42]. For example, lysine can be acylated by acyl-CoAs formed by the breakdown of fatty acids, and this mostly occurs through non-enzymatic mechanisms. Interestingly, some acyl-CoAs, such as glutaryl coenzyme A and succinyl coenzyme A, are derived from the carboxy cycle process but are more active than acetyl-CoA. The primary mechanism underpinning these modifications is acyl-CoA carboxyl group-induced intramolecular nucleophilic attack on CoA thioester bonds, which results in the formation of cyclic anhydride. This is more active than acyl-CoA and can produce non-enzymatic modifications more efficiently. Non-enzymatic acylation is thought to be affected by the cellular concentration of acyl-CoAs, the reactivity of acyl-CoA, local pH levels, and the number of lysine residues in proteins. These factors may differ between cell and tissue types. However, the specific regulatory mechanisms of this non-enzymatic modification remain unclear and require further investigation.
2. Acetylation in the DDR
2.1. Histone Acetylation in the DDR
Histone acetylation is well known to lead to changes in the structure of chromatin. Mechanistically, this occurs in two ways: on the one hand, the positive charge of lysine residues can be neutralized after acetylation, resulting in the weakening of the interaction between histone and DNA skeleton and the promotion of chromatin decompaction. This exposes more sites and enhances the accessibility of nucleosomal DNA. On the other hand, related chromatin remodeling complexes, such as SWI/SNF complexes, can also be recruited to the chromatin region to regulate chromatin structure. It was found that, in response to ionizing radiation (IR), nuclear ACLY is phosphorylated at S455 by ataxia telangiectasia mutated (ATM) and facilitates histone acetylation at DSBs, promoting HR-mediated repair by enabling BRCA1 recruitment while impairing 53BP1 recruitment [43]. This direct evidence demonstrates the link between histone acetylation and DDR. Other studies have shown that the acetylation modification of histone H1K85 can mediate chromatin changes under the dynamic regulation of acetylase PCAF and deacetylase HDAC1 in response to DNA damage, thus ensuring genome stability [44]. However, compared with H1 histone, H2 histone acetylation has been studied more extensively in terms of DDR. When cells are stimulated by ionizing radiation (IR), H2AX lysine 36 (H2AX K36ac) can be acetylated by acetyltransferase CBP/P300 to recruit corresponding DNA repair proteins to DNA damage sites [45]. Furthermore, under ionizing radiation-induced stimulation, the DNA PKCs BRD domain can specifically recognize H2AX acetylated lysine 5 (K5ac), which can lead to the determination of cell fate via γH2AX [46]. Moreover, under IR stimulation, knockdown of the tumor suppressor gene ZNF668, known to be involved in breast cancer, will weaken the interaction between Tip60 and H2AX. This leads to reduced hyperacetylation of histone H2AX and the prevention of chromatin relaxation, resulting in a reduced recruitment of repair proteins to DNA damage sites, defective homologous recombination (HR) repair, and reduced cell survival rates. These examples fully illustrate the importance of H2 histone acetylation to the DDR and, thus, to cancer [47].
Histone H3 acetylation also serves an important regulatory role in the DDR. At the location of DNA damage, the histone deacetylases HDAC1 and HDAC2 maintain low H3K56 acetylation levels to ensure that the damaged DNA is repaired. After the repair is completed, the proteasome will degrade the acetylated core histones, and the newly formed core histones will be assembled into nucleosomes. This represents the completion of the process of coping with DNA damage and repair [48]. PhD bromo tandem domain containing trimer motif 66 (TRIM66) has also been found to recognize unmodified H3R2, H3K4, and acetylated H3K56, while TRIM66 can recruit SIRT6 to deacetylate H3K56, thereby initiating the DDR and maintaining genome stability [49]. In addition, after 24 h of stimulation of HaCaT cells with sodium arsenite (NaAsO2), arsenic has been reported to reduce histone H3K18 acetylation levels, affect the expression of xeroderma pigmentosa-related proteins (XPA, XPD, and XPF nucleotide excision repair (NER)-related genes), and further aggravate DNA damage. However, the use of histone deacetylase inhibitor trichostatin A (TSA) can inhibit the deacetylation of H3K18 in the promoter region of XPA, XPD, and XPF, increase the acetylation of H3K18, and promote the transcriptional expression of NER-related genes. To a certain extent, it can inhibit arsenic-induced DNA damage [50].
In addition to H1, H2, and H3 acetylation, some studies also have been conducted on histone H4 acetylation. After doxorubicin treatment, cells in G0/G1 phase experience DNA damage but it can also promote the activation of chromatin kinase VRK1. VRK1 directly interacts with Tip60 and phosphorylates it, resulting in increased histone H4K16 acetylation, a marker of local chromatin relaxation. Inhibition of Tip60 expression by siRNA or its kinase inhibitor MG149 inhibited H4K16 acetylation, indicating VRK1-mediated phosphorylation of Tip60 increases its enzymatic activity. This work suggests that the dynamic remodeling of chromatin is closely related to the epigenetic modification of histone [51]. In addition, males absent of the first (MOF) proteins can regulate the level of H4K16 acetylation. MOF is responsible for maintaining sufficient levels of H4K16 acetylation in cells, which facilitates the generation of chromatin structures conducive to DNA repair. When MOF is absent in cells, the acetylation level of H4K16 is reduced and ultimately, the recruitment and cancellation of the corresponding signal proteins at the DNA damage site is impaired [52]. Furthermore, after DNA double-strand breaks induced by the HO endonuclease system, if the acetylation sites of newly synthesized histone H4 are mutated, the reassembly of chromatin structure is inhibited. Interestingly, the newly synthesized histone H4 acetylation mutation site changes, resulting in phosphorylated H2A (γ-H2AX) levels significantly decreasing around DSBs, indicating the critical role of chromatin assembly in DNA damage signaling [53].
In summary, Kac modification of histone tails can lead to the relaxation of chromatin structure, which is more conducive to the completion of DNA repair after DNA damage. Moreover, after DNA repair is completed, histones undergo deacetylation, and the chromatin structure becomes compact. Thus, this dynamic regulation by histone acetylation is critical to the maintenance of genome stability.
2.2. Non-Histone Acetylation in the DDR
Non-histone acetylation has also been found to participate in the DDR process. When inducing DNA damage in human cells, the acetylation of lysine 382 and phosphorylation of serine 392 in p53, a key DDR factor, can significantly enhance the interaction between p53 and MDC1 and promote the recruitment of these two proteins to DNA damage sites [54]. In response to DNA damage, N-acetyltransferase 10, NAT10 (also known as HALP), a member of the GNAT family, translocates to the nucleoplasm, promoting p53 acetylation at K120 with its acetyltransferase activity and proteosome-mediated degradation of MDM2 with its intrinsic E3 ligase activity, ultimately resulting in stabilizing p53 and p53-mediated cell cycle arrest and apoptosis [55]. Tip60-mediated acetylation, the DDR core kinase ATM also activates its kinase activity and subsequent checkpoint signaling upon DNA damage, while Tip60 inactivation sensitizes cells to ionizing radiation [56].
Werner syndrome is a rare autosomal recessive disease caused by mutations of the WRN gene. When the lysines K1127 and K1117 of WRN are mutated to arginine, cells may become sensitive to DNA-damaging agents such as mitomycin C and etoposide, indicating defective DNA repair. In fact, these two sites are critical for the recruitment of WRN to DNA damage sites [57].
Furthermore, the acetylation of proteins has also been found to be involved in the regulation of base excision repair (BER). For example, acetylation of depuridine/depyrimidine endonuclease 1 (APE1; also known as APEX1), an important regulator of BER, inhibits its interaction with XRCC1 when DNA damage occurs; resulting in decreased APE1 activity. However, SIRT1 can deacetylate APE1, recovering its function [58]. Additional acetylated non-histone proteins involved in the DDR are summarized in Table 1.
2.3. Roles of HATs and HDACs/SIRTs in DDR
Many HATs-mediated acetylations of histone and non-histone proteins directly or indirectly modulate DDR. KAT8 (hMOF), as a member of the histone acetyltransferase MYST family, can respond to DNA damage by catalyzing the acetylation of H4 at K16 (H4K16) and p53 at K120 [59,60]. KAT5 (TIP60) is also a member of the histone acetyltransferase MYST family [61], and its regulatory role in DNA damage signaling [62] has been reported. It was found that TIP60 can regulate the acetylation of a variety of histones (H2, H3, and H4) and non-histones (p53 [63] and ATM [64]). For example, TIP60 can acetylate the K15 position of H2A in response to DNA damage [65,66]. It has also been shown that Tip60 regulates the acetylation of H4 by forming a complex with transformation/transfer domain-associated protein (Trrap) and then promotes DNA damage repair by HR [67]. In addition, histone acetyltransferases KAT3A (CBP) and KAT3B (p300) are structurally similar and participate in the regulation of many functions in cells. In vitro enzyme activity test showed that CBP/p300 could acetylate all acetylation residue sites of histone H2A and H2B, among which K14, K18, and K56 of H3 and K5 and K8 of H4 were preferentially oxidized [68]. Moreover, CBP can also regulate the acetylation of non-histones in cancer. In human colon cancer cells, CBP can mediate the acetylation of K358 of DOT1L, which is positively correlated with the staging of colon cancer [69]. It was found that in vitro, acetyltransferase GCN5 can form a complex with its chaperone PCAF to acetylate multiple lysine residue sites on histone H3, such as H3K9, H3K14, H3K18, and H3K23 [70,71,72,73]. In addition, PCAF can acetylate H1K85 in response to DNA damage [74].
On the other hand, HDACs/SIRTs-mediated protein deacetylation also plays an important role in DDR. In mammals, SIRT1-7 has different subcellular localization, functions, and substrates and was initially identified as histone and non-histone protein deacetylases [75,76]. It was found that sirtuins can mediate the specific deacetylation of histone lysine residues to facilitate DNA repair. In mammals, H3K56 will undergo hyperacetylation upon inhibition of SIRT1 expression, leading to the instability of the S phase genome [77]. It has also been reported thatSIRT3, which is mainly located in mitochondria, will be transported to the nucleus to regulate the deacetylation of H4K16ac in response to DNA damage [78,79,80]. SIRT6 is an intranuclear deacetylase which plays an important role in regulating DDR signal and genome stability. Its histone substrates include H3K9, H3K18, and H3K56. H3K9 deacetylation mediated by SIRT6 can protect telomeres in mammalian cells. On the contrary, the lack of SIRT6 will lead to chromosome fusion due to telomere dysfunction [81]. Similarly, in the nucleus, SIRT7 can catalyze the deacetylation of H3K18ac at the late response to IR [82]. In addition, HDAC1/2-mediated deacetylation of H3K56 and H4K16 also plays an important role in chromatin regulation [83,84]. It was found that histone deacetylase was rapidly recruited to the DNA damage site, leading to histone deacetylation, thereby promoting non-homologous end joining (NHEJ) repair [83]. Several studies have found that the levels of H3K56ac, H4K16ac, and H4K91ac will increase after HDAC1 expression is inhibited, which is related to the decreased cell survival rate after treatment with DNA damaging reagents [83,84,85,86].
In conclusion, the dynamic regulation of both histone and non-histone acetylation and deacetylation serves an important function in the repair of DNA damage.
Table 1. Lipid metabolite associated PTMs in the DDR.
| Modification | Writers | Erasers | Readers | Substrates in the DDR | References |
|---|---|---|---|---|---|
| Acetylation (histone) | P160, P300/CBP | HDAC1-11, | YAF9, ENL, AF9, Taf14, |
H1K85 | |
| TAFII230, MYST, | SIRT1-7 | Sas5(Yeats), | H2AX K5, K36 | [44,45,46,49,50,51] | |
| GNAT, PCAF | PHDs | H3K4, K18, K56 H4K16 |
|||
| Acetylation (non-histone) |
GCN5, P300, MYST19, | HDAC1-11 | Tip60, APE1, | ||
| KAT1, TAT1, ESCO1-2 | SIRT1-7, LEF1, TCF1 | NA | PFKFB3 K472, OGG1, Cdc25A, P53K382/K120, WRN K1117/K1127, MLH1, RRM2 PARP1 K949, |
[34,87,88,89,90,91] | |
| Succinylation | NA | KDAC: SIRT5, SIRT7 | NA | H3K12, P53K120, H4K77, | |
| ACOX1(acyl-CoA oxidase 1), FEN1 K200, NPM1 | [19,92,93,94,95,96] | ||||
| Palmitoylation | DHHC1-23 | APT1-2 | NA | Rap1-interacting factor 1(Rif1) C466/C473 | [97] |
| N-myristoylation | NMT1-2 | SIRT1-3, SIRT6 | NA | Finkel-Biskis-Reilly (FBR) v-fos | [98] |
| Crotonylation | P300/CBP, MOF | HDAC1-3, SIRT1-3 | YEATS, PHD | RPA1 | [21] |
Abbreviations: NA, not available; ESCO1, the establishment of cohesion 1 homolog 1; PFKFB3, phosphofructokinase-2/fructose-2,6-bisphosphatase 3; WRN, Werner syndrome protein; MLH1, MutL Homolog 1; RRM2, Ribonucleoside-Diphosphate Reductase Subunit M2; FEN-1, flap endo/exonuclease; RPA1, Replication factor A protein 1; KAT, lysine (K) acetyltransferase family; DHHC, aspartate–histidine–histidine–cysteine family; NMT, N-terminal myristoyltransferase family.
3. Acetylation in Cancer
3.1. HATs and Cancer
HATs are well known to be involved in tumorigenesis and cancer progression, with HAT activity being altered either by gene mutations or viral oncogenes in both blood and solid cancers. For example, the interaction between adenovirus SV40T antigen protein E1A and the co-activators p300 and CBP play a key role in cell transformation [99]. These HATs are then redistributed to the promoter regions of certain genes to promote cell growth, differentiation, and the transcriptional activation of specific genes [100]. Ambiguous p300 mutations can be found in solid stomach, rectum, breast, and prostate tumors [101,102,103].
Tip60 is another HAT that is closely associated with tumorigenesis [104,105,106] and may be involved in the regulation of DNA repair and the transcriptional activation of p53 and Myc [62,107]. Decreased expression of Tip60 results in low p53 acetylation levels and incomplete apoptosis signaling, which indicates transformation to a malignant tumor [108]. As a tumor suppressor protein, single allele deletion of human Tip60 is often found in head and neck tumors, breast cancer, and lymphoma [109]. In addition, Tip60 has also been found to inhibit Myc-mediated lymphoma formation in B-cell lymphoma [110].
3.2. HDACs and Cancer
HDACs function in the opposite manner to HATs, regulating transcription by removing acetyl groups from lysine residues of histone tails and other non-histone substrates. Thus, it stands to reason that they would also be involved in cancer. Functional experiments have indicated that type I HDACs are mainly responsible for regulating cell proliferation and apoptosis, while type II HDACs regulate cell metastasis and angiogenesis. For example, inhibiting HDAC1 and HDAC2 in vitro can inhibit colon cancer cell proliferation [111,112]. However, the inhibition of HDAC3 suppresses the proliferation of colon cancer cells more substantially [113]. In addition, inhibition of HDAC2 and HDAC3 is known to induce DDR and apoptosis after DNA damage.
Type II and IV HDACs are primarily localized in the cytoplasm and are mainly responsible for the deacetylation of non-histone proteins. Previous studies have shown that inhibiting HDAC4 reduces colon cancer cell proliferation and induces apoptosis [114,115,116]. Furthermore, while the inhibition of HDAC7 in endothelial cells does not affect cell growth and survival, it does inhibit cell metastasis and the formation of capillary-like structures in cancer [117,118]. Type II HDACs are mainly responsible for regulating angiogenesis, and inhibition of HDAC6 and HDAC10 results in decreased VEGFR1 and two expressions [119].
Type III HDACs, also known as the sirtuins, share no sequence homology with other deacetylases. However, they may also be involved in regulating the occurrence and development of tumors. Sirtuins induce the deacetylation of a range of protein substrates, including histones, but also mediate ADP ribosylation. Furthermore, overexpression of SIRT1, 2, 3, and 7 have been identified in many types of cancer [120,121,122]. For example, overexpression of SIRT1 is known to prevent apoptosis by regulating histone deacetylation, promoter methylation, and histone methylation and inhibiting the transcription of tumor suppressor genes. This ultimately promotes cancer cell growth by preventing cell senescence and differentiation, as well as the formation of tumor blood vessels by promoting the growth of endothelial cells and preventing their aging [123]. Interestingly, the expression of SIRT2 is absent in human glioma cells, and re-expression of SIRT2 can reduce the ability of colony-stimulating factor formation of cells [124]. This suggests that, in some cases, Sirtuins also serve as tumor suppressors. At present, whether Sirtuins function as oncogenes or as tumor suppressors remains controversial. However, it is clear that altered HDAC function plays a corresponding role to that of HATs in the process of tumorigenesis and development.
3.3. Summary
The extensive research conducted thus far on protein acetylation and the fact that abnormal acetylation is closely associated with cancer has laid the foundation for the discovery of many novel epigenetic drug targets. Certain HDAC inhibitors have been approved for cancer treatment, including romidepsin, panobinostat, and belinostat, while more are being tested in clinical trials. However, most research exploring the potential for HAT inhibitors to treat cancer is yet to enter clinical trials, leaving this treatment avenue in its primary stages. However, the identification of new HAT subtypes and improved characterization of their roles and functions have provided more potential treatment strategies. For example, curcumin, as a natural KATi, can inhibit the activity of p300/CBP and so suppress the proliferation of a variety of cancer cells, thereby achieving anti-inflammatory and anti-tumor effects [125]. Moreover, as a small molecule derived from anacardic acid, MG153 also acts as a potential p300/PCAF inhibitor and can suppress the proliferation of BCR-ABL-expressing cells, induce apoptosis, and resist DNA damage [126]. In addition, L002, a small molecule inhibitor of p300, has been shown to inhibit p300, PCAF, and GCN5 activity in leukemia, lymphoma, and breast cancer cell lines [127]. In conclusion, further research on both HDAC and HAT inhibitors will likely prove very fruitful when developing novel treatments for cancer [128].
This entry is adapted from the peer-reviewed paper 10.3390/biom12111655
This entry is offline, you can click here to edit this entry!