Fluorine forms a great variety of chemical compounds, within which it always adopts an oxidation state of −1. With other atoms, fluorine forms either polar covalent bonds or ionic bonds. Most frequently, covalent bonds involving fluorine atoms are single bonds, although at least two examples of a higher order bond exist. Fluoride may act as a bridging ligand between two metals in some complex molecules. Molecules containing fluorine may also exhibit hydrogen bonding (a weaker bridging link to certain nonmetals). Fluorine's chemistry includes inorganic compounds formed with hydrogen, metals, nonmetals, and even noble gases; as well as a diverse set of organic compounds.[note 1] For many elements (but not all) the highest known oxidation state can be achieved in a fluoride. For some elements this is achieved exclusively in a fluoride, for others exclusively in an oxide; and for still others (elements in certain groups) the highest oxidation states of oxides and fluorides are always equal.
- fluorine atoms
- fluorine
- fluorides
1. Difluorine
While an individual fluorine atom has one unpaired electron, molecular fluorine (F2) has all the electrons paired. This makes it diamagnetic (slightly repelled by magnets) with the magnetic susceptibility of −1.2×10−4 (SI), which is close to theoretical predictions.[1] In contrast, the diatomic molecules of the neighboring element oxygen, with two unpaired electrons per molecule, are paramagnetic (attracted to magnets).[2]
| X | XX | HX | BX3 | AlX3 | CX4 |
|---|---|---|---|---|---|
| F | 159 | 574 | 645 | 582 | 456 |
| Cl | 243 | 428 | 444 | 427 | 327 |
| Br | 193 | 363 | 368 | 360 | 272 |
| I | 151 | 294 | 272 | 285 | 239 |
The fluorine–fluorine bond of the difluorine molecule is relatively weak when compared to the bonds of heavier dihalogen molecules. The bond energy is significantly weaker than those of Cl2 or Br2 molecules and similar to the easily cleaved oxygen–oxygen bonds of peroxides or nitrogen–nitrogen bonds of hydrazines.[4] The covalent radius of fluorine of about 71 picometers found in F2 molecules is significantly larger than that in other compounds because of this weak bonding between the two fluorine atoms.[5] This is a result of the relatively large electron and internuclear repulsions, combined with a relatively small overlap of bonding orbitals arising due to the small size of the atoms.[6]
The F2 molecule is commonly described as having exactly one bond (in other words, a bond order of 1) provided by one p electron per atom, as are other halogen X2 molecules. However, the heavier halogens' p electron orbitals partly mix with those of d orbitals, which results in an increased effective bond order; for example, chlorine has a bond order of 1.12.[7] Fluorine's electrons cannot exhibit this d character since there are no such d orbitals close in energy to fluorine's valence orbitals.[7] This also helps explain why bonding in F2 is weaker than in Cl2.[6]
1.1. Reactivity
Reactions with elemental fluorine are often sudden or explosive. Many substances that are generally regarded as unreactive, such as powdered steel, glass fragments, and asbestos fibers, are readily consumed by cold fluorine gas. Wood and even water burn with flames when subjected to a jet of fluorine, without the need for a spark.[8][9]
Reactions of elemental fluorine with metals require different conditions that depend on the metal. Often, the metal (such as aluminium, iron, or copper) must be powdered because many metals passivate by forming protective layers of the metal fluoride that resist further fluoridation.[3] The alkali metals react with fluorine with a bang (small explosion), while the alkaline earth metals react not quite as aggressively. The noble metals ruthenium, rhodium, palladium, platinum, and gold react least readily, requiring pure fluorine gas at 300–450 °C (575–850 °F).[10]
Fluorine reacts explosively with hydrogen in a manner similar to that of alkali metals.[11] The halogens react readily with fluorine gas[12] as does the heavy noble gas radon.[13] The lighter noble gases xenon and krypton can be made to react with fluorine under special conditions, while argon will undergo chemical transformations only with hydrogen fluoride.[14] Nitrogen, with its very stable triple bonds, requires electric discharge and high temperatures to combine with fluorine directly.[15] Fluorine reacts with ammonia to form nitrogen and hydrogen fluoride .
2. Chemical Characteristics, Effects of Presence in a Molecule
Fluorine's chemistry is dominated by its strong tendency to gain an electron. It is the most electronegative element and elemental fluorine is a strong oxidant. The removal of an electron from a fluorine atom requires so much energy that no known reagents are known to oxidize fluorine to any positive oxidation state.[16]
Therefore, fluorine's only common oxidation state is −1. It differs from this value in elemental fluorine, where the atoms are bonded to each other and thus at oxidation state 0, and a few polyatomic ions: the very unstable anions F−2 and F−3 with intermediate oxidation states exist at very low temperatures, decomposing at around 40 K.[17] Also, the F+4 cation and a few related species have been predicted to be stable.[18]
Fluorine forms compounds with all elements except neon and helium. In particular, it forms binary compounds, named fluorides, with all said elements except argon. All of the elements up to einsteinium, element 99, have been checked except for astatine and francium,[19] and fluorine is also known to form compounds with mendelevium, element 101,[20] rutherfordium, element 104,[21] and seaborgium, element 106.[22]
As a result of its small size and high negative charge density, the fluoride anion is the "hardest" base (i.e., of low polarizability). Because of this, fluorides in real salt crystals often have higher effective charges than oxides of the same metal, even though oxygen's formal charge is twice as great as fluorine's.
As a part of a molecule, it is a part with great inductive effect. In the latter case, it significantly increases the acidity of a molecule: the anion formed after giving the proton off becomes stable as a result. Consider acetic acid and its mono-, di-, and trifluoroacetic derivatives and their pKa values (4.74, 2.66, 1.24, and 0.23[23]);[24] in other words, the trifluoro derative is 33,800 times stronger an acid than acetic.[25] Fluorine is a principal component of the strongest known charge-neutral acid, H2FSbF6.[26] There is evidence for an even stronger acid, H2FAuF6, but it has not proved isolable.[27]
In a molecule that is composed of a central atoms and fluorines attached to it, the intermolecular bonding is not very strong. Moreover, the dense negative balls that fluorines are repel each other. Therefore, the intermolecular bonding strength falls further down, a result of which is the low melting point of high fluorides.
3. Hydrogen Fluoride
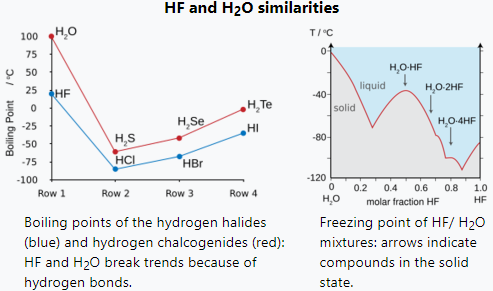
Fluorine combines with hydrogen to make a compound (HF) called hydrogen fluoride or, especially in the context of water solutions, hydrofluoric acid. The H-F bond type is one of the few capable of hydrogen bonding (creating extra clustering associations with similar molecules). This influences various peculiar aspects of hydrogen fluoride's properties. In some ways the substance behaves more like water, also very prone to hydrogen bonding, than one of the other hydrogen halides, such as HCl.[28][29][30]
Hydrogen bonding amongst HF molecules gives rise to high viscosity in the liquid phase and lower than expected pressure in the gas phase. Hydrogen fluoride does not boil until 20 °C in contrast to the heavier hydrogen halides which boil between −85 °C and −35 °C (−120 °F and –30 °F). HF is miscible with water (will dissolve in any proportion), while the other hydrogen halides have large solubility gaps with water. Hydrogen fluoride and water also form several compounds in the solid state, most notably a 1:1 compound that does not melt until −40 °C (−40 °F), which is 44 degrees Celsius (79 degrees Fahrenheit) above the melting point of pure HF.[31]
Unlike other hydrohalic acids, such as hydrochloric acid, hydrogen fluoride is only a weak acid in water solution, with acid dissociation constant (pKa) equal to 3.19.[32] HF's weakness as an aqueous acid is paradoxical considering how polar the HF bond is, much more so than the bond in HCl, HBr, or HI. The explanation for the behavior is complicated, having to do with various cluster-forming tendencies of HF, water, and fluoride ion, as well as thermodynamic issues.[33] At great concentrations, a property called homoconjugation is revealed. HF begins to accept fluoride ions, forming the polyatomic ions (such as bifluoride, HF−2) and protons, thus greatly increasing the acidity of the compound.[34] Hydrofluoric acid is also the strongest of the hydrohalic acids in acetic acid and similar solvents.[35] Its hidden acidity potential is also revealed by the fact it protonates acids like hydrochloric, sulfuric, or nitric.[36] Despite its weakness, hydrofluoric acid is very corrosive, even attacking glass (hydrated only).[34]
Dry hydrogen fluoride dissolves low-valent metal fluorides readily. Several molecular fluorides also dissolve in HF. Many proteins and carbohydrates can be dissolved in dry HF and can be recovered from it. Most non-fluoride inorganic chemicals react with HF rather than dissolving.[37]
4. Metal Fluorides
Metal fluorides are rather dissimilar from other metal halides, adopting distinctive structures. In many respects, metal fluorides are more similar to oxides, often having similar bonding and crystal structures.[38]
Owing to its high electronegativity, fluorine stabilizes metals in higher oxidation states with high M:halide ratios. Numerous charge-neutral penta- and hexafluorides are known, whereas analogous chlorides and bromides are rarer. The molecular binary fluorides are often volatile, either as solids [39] liquids,[40] or gases[41] at room temperature.
The solubility of fluorides varies greatly but tends to decrease as the charge on the metal ion increases. Dissolved fluorides produce basic solutions.[42]

4.1. Low Oxidation State Metal Fluorides
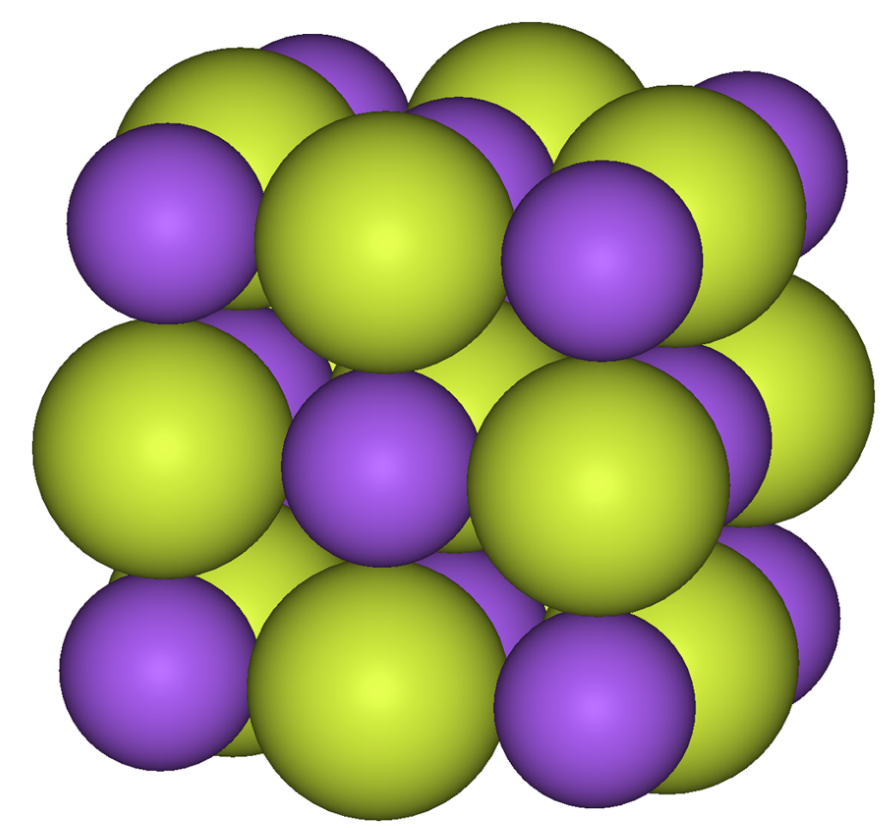
The alkali metals form monofluorides. All are soluble and have the sodium chloride (rock salt) structure,[43] Because the fluoride anion is basic, many alkali metal fluorides form bifluorides with the formula MHF2. Among other monofluorides, only silver(I)[44] and thallium(I)[45] fluorides are well-characterized. Both are very soluble, unlike the other halides of those metals.
Unlike the monofluorides, the difluorides may be either soluble or insoluble. Several transition metal difluorides, such as those of copper(II) and nickel(II), are soluble.[44] The alkaline earth metals form difluorides that are insoluble.[44] In contrast, the alkaline earth chlorides are readily soluble.[44]
Many of the difluorides adopt the fluorite structure, named after calcium fluoride (and also adopted by several metal dioxides such as CeO2, UO2, ThO2, etc.), which surrounds each metal cation with 8 fluorides. Some difluorides adopt the rutile structure, named after a form of titanium dioxide and adopted by several other metal dioxides also. The structure is tetragonal and puts metal atoms in octahedral coordination.
Beryllium difluoride is different from the other difluorides. In general, beryllium has a tendency to bond covalently, much more so than the other alkaline earths and its fluoride is partially covalent (although still more ionic than its other halides). BeF2 has many similarities to SiO2 (quartz) a mostly covalently bonded network solid. BeF2 has tetrahedrally coordinated metal and forms glasses (is difficult to crystallize). When crystalline, beryllium fluoride has the same room temperature crystal structure as quartz and shares many higher temperatures structures also.
Beryllium difluoride is very soluble in water,[44] unlike the other alkaline earths. (Although they are strongly ionic, they do not dissolve because of the especially strong lattice energy of the fluorite structure.) However, BeF2 has much lower electrical conductivity when in solution or when molten than would be expected if it were ionic.[46][47][48][49]

Many metals form trifluorides, such as iron, bismuth, the rare-earth elements, and the metals in the aluminium and scandium columns of the periodic table. The trifluorides of many rare earths, as well as bismuth, have the YF3 structure. Trifluorides of plutonium, samarium (at high temperature), and lanthanum adopt the LaF3 structure. Iron and gallium trifluorides have the FeF3 structure, which is similar to rhenium trioxide. Only ScF3 is cubic (ReO3) at ambient temperature; this material also has the unusual property of negative thermal expansion, meaning it shrinks on heating, over a quite broad temperature range.
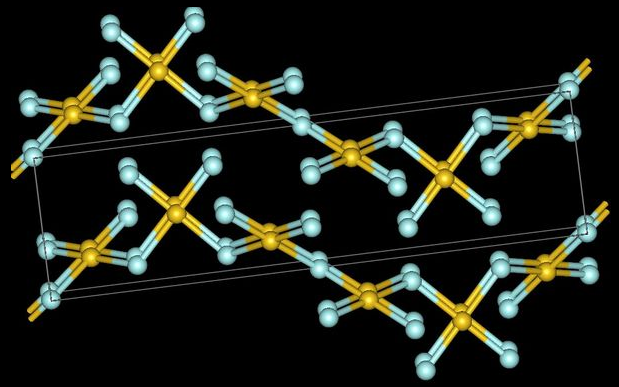
Gold trifluoride adopts a structure of linked –AuF4– squares that align in a helix (spiral chain).[50] In contrast to gold's distinctly ionic trifluoride, its trichloride and tribromide are volatile dimeric molecules.[51] Aluminium trifluoride is a high melting point solid which is a monomer in the gas phase, while its other trihalides are low-melting, volatile molecules or linear polymeric chains that form dimers as gases phase.[52] No trifluoride is soluble in water, but several are soluble in other solvents.[53]
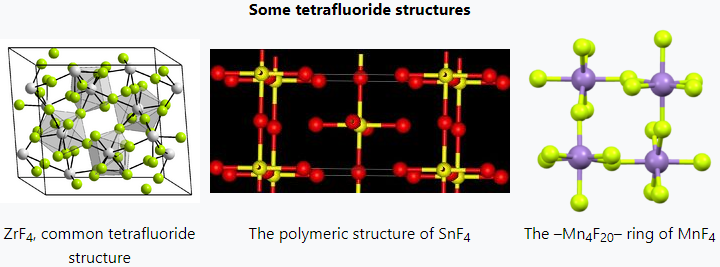
The tetrafluorides show a mixture of ionic and covalent bonding. Zirconium, hafnium, plus many of the actinides form tetrafluorides with an ionic structure that puts the metal cation in an 8-coordinate square antiprism.[54][55] Melting points are around 1000 °C.[56]
Titanium and tin tetrafluorides are polymeric, with melting points below 400 °C. (In contrast, their tetrachlorides are molecular and liquids at room temperature.) Vanadium tetrafluoride has a similar structure to tin's[57] and disproportionates at 100–120 °C to the trifluoride and the pentafluoride.
The tetrafluorides of iridium, platinum, palladium, and rhodium all share the same structure which was not known until 1975. They have octahedrally coordinated metal atoms with four of the fluorines shared and two unshared. The melting points, where known, are below 300 °C.
Manganese tetrafluoride is an unstable solid that decomposes even at room temperature.[58] Only one of the two allotropes, α-MnF4, is understood. In this compound, manganese forms –MnF6– octahedra which share bridging fluorines to make –Mn4F20– rings which are then further connected three dimensionally.[59]
4.2. High Oxidation State Metal Fluorides
Metal penta- and higher fluorides are all molecular and hence at least somewhat volatile.
Vanadium, niobium, and tantalum form pentafluorides as their highest charge-neutral fluoride. Vanadium pentafluoride is the only non-volatile high-charged metal fluoride, with vanadium being centers of –VF6– octahedra. The niobium and tantalum pentafluorides, have the same tetrahedra in their structures, with the difference being the formation of the tetra- (rather than poly-) meric molecules.[60]
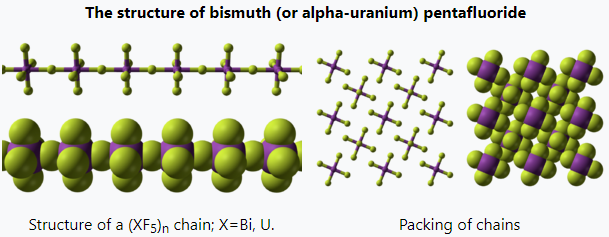
Bismuth's highest fluoride is a volatile penta species that is a powerful fluorinating agent. In the solid state, it is polymeric, consisting of linear chains of octahedra, sharing axial fluorides. In combination with alkali metals, pentavalent bismuth can form hexafluorobismuthate, [BiF6]−, upon reaction with a fluoride donor, either strong (such as NaF[61][62]) or not (such as XeF4[63]).
Many metals that form hexafluorides also can form pentafluorides. For instance, uranium, which has a well-known hexafluoride, also forms two different pentafluoride structures. The room-temperature (alpha) form has the same linear chain structure as bismuth pentafluoride. As a molecular (gas) species, UF5 has a square pyramidal structure.
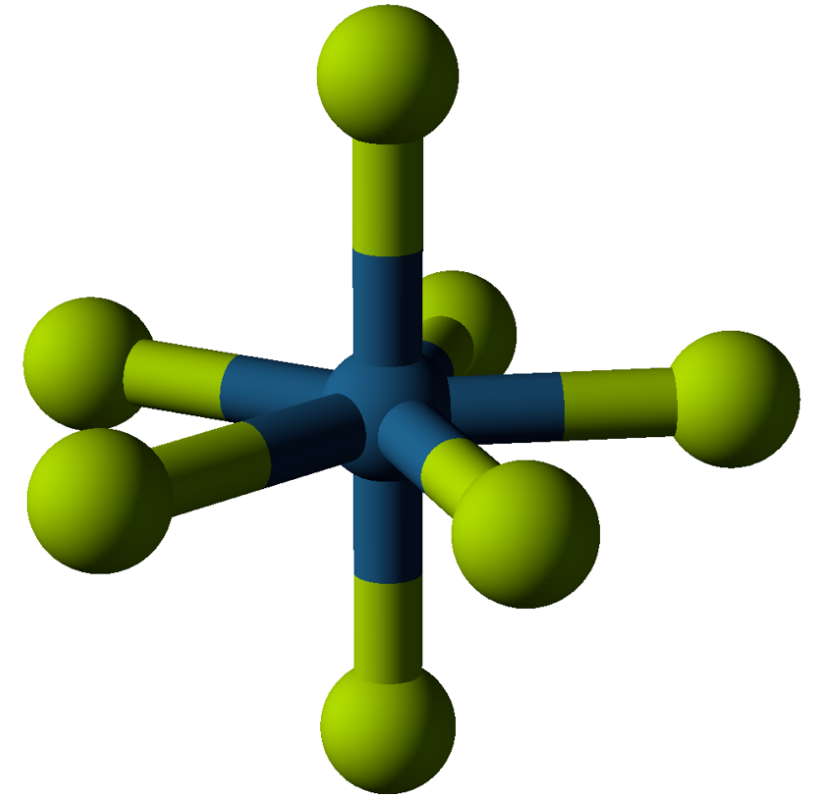
The metals that make well-characterized hexafluorides include nine metals in the center of the periodic table (molybdenum, technetium, ruthenium, rhodium, tungsten, rhenium, osmium, iridium, and platinum) along with elements 92–94: uranium, neptunium, and plutonium. At room temperature, tungsten hexafluoride is a gas. Molybdenum hexafluoride and rhenium hexafluoride are liquids. The rest are volatile solids.

Metal hexafluorides are oxidants because of their tendency to release fluorines: for example, platinum hexafluoride was the first compound to oxidize molecular oxygen[64] and xenon.[65] Polonium also forms a hexafluoride, but it is understudied.[66]
Rhenium is the only metal known to bond with seven fluorides, which is the record for number of charged ligands for a charge-neutral metal compound.[67]Rhenium heptafluoride adopts a pentagonal bipyramid molecular geometry. Calculations shows that the currently unknown but perhaps possible iridium heptafluoride[68] (report of synthesis is being prepared[69]), technetium heptafluoride,[70] and osmium heptafluoride[71] will also have this structure).
Osmium octafluoride was first reported in 1913, but in 1958 that compound was shown to be actually osmium hexafluoride.[72] A 1993 theoretical study predicted very weak bonds in osmium octafluoride and said that it would be difficult to ever detect experimentally. The study predicted that, if made, OsF8 would have Os–F bonds of two different lengths.[73]
5. Nonmetal Fluorides
The nonmetal binary fluorides are volatile compounds. They show a great difference between period 2 and other fluorides. For instance, period 2 elements elements fluorides never exceed the octet in their atoms. (Boron is an exception due to its specific position in the periodic table.) Lower-period elements, however, may form hypervalent molecules, such as phosphorus pentafluoride or sulfur hexafluoride.[74] The reactivity of such species varies greatly—sulfur hexafluoride is inert, while chlorine trifluoride is extremely reactive—but there are some trends based on periodic table locations.
Boron trifluoride is a planar molecule. It has only six electrons around the central boron atom (and thus an incomplete octet), but it readily accepts a Lewis base, forming adducts with lone-pair-containing molecules or ions such as ammonia or another fluoride ion which can donate two more electrons to complete the octet.[75] Boron monofluoride is an unstable molecule with an unusual (higher than single) bond to fluorine. The bond order has been described as 1.4 (intermediate between a single and double bond). It is isoelectronic with N2.[76]

Silicon tetrafluoride, similar to carbon tetrafluoride and germanium tetrafluoride, adopts a molecular tetrahedral structure.[77] SiF4 is stable against heating or electric spark, but reacts with water (even moist air), metals, and alkalies, thus demonstrating weak acidic character.[78] Reactions with organomagnesium compounds, alcohols, amines, and ammonia yield adduction compounds.[78] Fluorosilicic acid, a derivative of SiF4, is a strong acid in aqueous solution (the anhydrous form does not exist).[79]
Pnictogens (nitrogen's periodic table column) show very similar trends in reactivity and acidity of the highest fluorides (pentafluorides) and most common ones (trifluorides), with the said property increasing down the group: NF3 is stable against hydrolysis,[80] PF3 hydrolyzes very slowly in moist air,[81] while AsF3 completely hydrolyzes.[80] SbF3 hydrolyzes only partially because of the increasing ionic character of the bond to fluorine. The compounds are weak Lewis bases, with NF3 again being an exception.[80] The pentafluorides of phosphorus[81] and arsenic[82] are much more reactive than their trifluorides; antimony pentafluoride is such a strong acid that it holds the title of the strongest Lewis acid.[82] Nitrogen is not known to form a pentafluoride, although the tetrafluoroammonium cation (NF+4) features nitrogen in the formal oxidation state of +5.[83] Nitrogen monofluoride is a metastable species that has been observed in laser studies. It is isoelectronic with O2 and, unusually, like BF, has a higher bond order than single-bonded fluorine.[84][85]
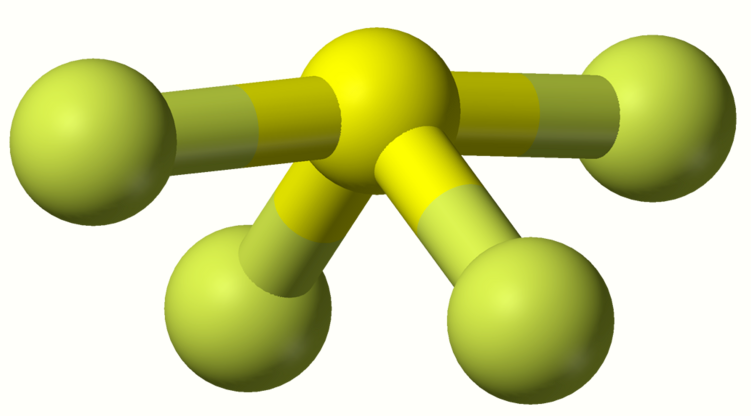
The chalcogens (oxygen's periodic table column) are somewhat similar: The tetrafluorides are thermally unstable and hydrolyze, and are also ready to use their lone pair to form adducts to other (acidic) fluorides. Sulfur and selenium tetrafluorides are molecular while TeF4 is a polymer.[86] The hexafluorides are the result of direct fluorination of the elements (compare: other hexahalides of these elements do not even exist). They increase in reactivity with atomic number: SF6 is extremely inert, SeF6 is less noble (for example, reacts with ammonia at 200 °C (400 °F)), and TeF6 easily hydrolyzes to give an oxoacid.[86] Oxygen's highest fluoride is oxygen difluoride,[86] but fluorine can theoretically (as of 2012) oxidize it to a uniquely high oxidation state of +4 in the fluorocation: OF+3.[87] In addition, several chalcogen fluorides occur which have more than one chalcogen (O2F2,[88] S2F10,[89] etc.).
The well-characterized heavier halogens (chlorine, bromine, and iodine) all form mono-, tri-, and pentafluorides: XF, XF3, and XF5. Of the neutral +7 species, only iodine heptafluoride is known.[90] While chlorine and bromine heptafluorides are not known, the corresponding cations ClF+6 and BrF+6, extremely strong oxidizers, are.[91] Astatine is not well-studied, and although there is a report of a non-volatile astatine monofluoride,[92] its existence is debated.[93] Many of the halogen fluorides are powerful fluorinators. Chlorine trifluoride is particularly noteworthy—readily fluorinating asbestos and refractory oxides—and may be even more reactive than chlorine pentafluoride. Used industrially, ClF3 requires special precautions similar to those for fluorine gas because of its corrosiveness and hazards to humans.[94][95]
5.1. Superacids
Several important inorganic acids contain fluorine. They are generally very strong because of the high electronegativity of fluorine. One such acid, fluoroantimonic acid (HSbF6), is the strongest charge-neutral acid known.[26] The dispersion of the charge on the anion affects the acidity of the solvated proton (in form of H2F+): The compound has an extremely low pKa of −28 and is 10 quadrillion (1016) times stronger than pure sulfuric acid.[26] Fluoroantimonic acid is so strong that it protonates otherwise inert compounds like hydrocarbons. Hungarian-American chemist George Olah received the 1994 Nobel Prize in chemistry for investigating such reactions.[96]
5.2. Noble Gas Compounds
The noble gases are generally non-reactive because they have filled electronic shells. Until the 1960s, no chemical bond with a noble gas was known. In 1962, Neil Bartlett found that platinum hexafluoride reacts with xenon. He called the compound he prepared xenon hexafluoroplatinate, but since then the product has been revealed to be mixture, perhaps monofluoroxenyl(II) pentafluoroplatinate, [XeF]+[PtF5]−, monofluoroxenyl(II) undecafluorodiplatinate, [XeF]+[Pt2F11]−, and trifluorodixenyl(II) hexafluoroplatinate, [Xe2F3]+[PtF6]−.[97] Bartlett's fluorination of xenon has been highly praised.[98] Later in 1962, xenon was found to react directly with fluorine to form the di- and tetrafluorides. Since then, other noble gas fluorides have been reported.
The binary compounds xenon include xenon difluoride, xenon tetrafluoride, and xenon hexafluoride.[99] Xenon forms several oxyfluorides, such as xenon oxydifluoride, XeOF2, by hydrolysis of xenon tetrafluoride.[100] Its lighter neighbor, krypton also forms well-characterized compounds, e.g., krypton difluoride. Krypton tetrafluoride was reported in 1963,[101] but was subsequently shown to be a mistaken identification; the compound seems to be very hard to synthesize now (although even the hexafluoride may exist).[102]
In accordance with the periodic trends, radon is more reactive toward fluorine. Radon difluoride has been claimed.[92] The lighter noble gases (helium through argon) do not form stable binary fluorides.
6. Highest Oxidation States: Fluorine Versus Oxygen
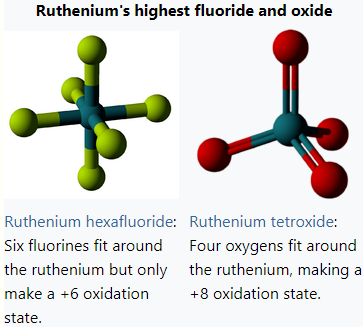
Elements frequently have their highest oxidation state in the form of a binary fluoride. Several elements show their highest oxidation state only in a few compounds, one of which is the fluoride; and some elements' highest known oxidation state is seen exclusively in a fluoride.
For groups 1–5, 10, 13–16, the highest oxidation states of oxides and fluorides are always equal. Differences are only seen in chromium, groups 7–9, copper, mercury, and the noble gases. Fluorination allows some elements to achieve relatively low[103] highest oxidation states that are otherwise hard to achieve. For example, no binary oxide is known for krypton, but krypton difluoride is well-studied.[104] At the same time, for some other elements, certain very high oxidation states are known only for the oxygen-based species, not the fluorine-based ones. For the previously mentioned volatile oxides, there are no corresponding hepta- or octafluorides. (For example, ruthenium octafluoride is unlikely to be ever synthesized,[105] while ruthenium tetroxide has even found an industrial use.[106]) The main problem that prevents fluorine from forming the highest states in covalent hepta- and octafluorides is that it is hard to attach such a large number of ligands around a single atom; the number of ligands is halved in analogous oxides.[107][108] However, octafluoride anions, such as the octafluoroiodate (IF−8), octafluorozirconate (ZrF4−8), and octafluoroxenate (XeF2−8) anions are well-known.
The highest oxidation states may be uncommon to everyday life, or even industrial usage. For example, the synthesis of mercury tetrafluoride, the first compound to achieve an oxidation state above +2 for a group 12 element, breaking the filled 5d-shell, again showing the significance of the relativistic effects on the heavy elements, and fueling the debate over whether mercury, cadmium, and zinc are transition metals,[109] occurred at cryogenic temperatures and the compound decomposes at the temperatures of solid nitrogen.[110] More unstable still, the only cobalt(V) species, the CoF+4 cation, has only been observed in gas phase (with no interactions with other atoms, thus no shown stability in any chemical environment).[105] The reason why such unstable species exist is complicated, yet can be summarized as follows on the example of the hypothesized NF5 molecule: According to the modern calculations, five fluorine atoms and one nitrogen atoms can theoretically arrange themselves in different ways, such as NF3 and F2, NF•4 and F•, NF5, etc. The NF3+F2 system is of the smallest energy (most stable). However, if a NF5 molecule was synthesized, it would have to go through a high-energy transitional state, from which it could decay into two molecules. But since the transitional state is higher in energy than the hexatomic molecule, the energy difference would need to be added to reach the transitional state and thus allow the decay. This energy is called reaction activation barrier. (The second decay mode has an analogical position.) Thus if little energy was added (low temperatures), then the compound could exist; however, the synthesis is a serious problem (not yet solved).[111]
7. Organofluorine Compounds
The carbon–fluorine chemical bond of the organofluorine compounds is the strongest bond in organic chemistry.[112] Along with the low polarizability of the molecules, these are the most important factors contributing to the great stability of the organofluorines.[113]
The carbon–fluorine bond of the smaller molecules is formed in three principal ways: Fluorine replaces a halogen or hydrogen, or adds across a multiple bond. The direct reaction of hydrocarbons with fluorine gas can be dangerously reactive, so the temperature may need to be lowered even to −150 °C (−240 °F).[114] "Solid fluorine carriers", compounds that can release fluorine upon heating, notably cobalt trifluoride,[115] may be used instead, or hydrogen fluoride. After the reaction, the molecular size is not changed significantly, as the elements have very similar van der Waals radii.[113] Direct fluorination becomes even less important when it comes to organohalogens or unsaturated compounds reactions, or when a prefluorocarbon is desired (then HF-based electrolysis is typically used).[116] In contrast, the fluoropolymers are formed by polymerizing free radicals; other techniques used for hydrocarbon polymers do not work in that way with fluorine.[117]
The range of organofluorine compounds is diverse, reflecting the inherent complexity of organic chemistry. A vast number of small molecules exist with varying amounts of fluorine substitution, as well as many polymers—research into particular areas is driven by the commercial value of applications.[118]

7.1. Small Molecules
Monofluoroalkanes (alkanes with one hydrogen replaced with fluorine) may be chemically and thermally unstable, yet are soluble in many solvents; but as more fluorines are in instead of hydrogens, the stability increases, while melting and boiling points, and solubility decrease. While the densities and viscosities are increased, the dielectric constants, surface tensions, and refractive indices fall.[119]
Partially fluorinated alkanes are the hydrofluorocarbons (HFCs). Substituting other halogens in combination with fluorine gives rise to chlorofluorocarbons (CFCs) or bromofluorocarbons (BFCs) and the like (if some hydrogen is retained, HCFCs and the like). Properties depend on the number and identity of the halogen atoms. In general, the boiling points are even more elevated by combination of halogen atoms because the varying size and charge of different halogens allows more intermolecular attractions.[120] As with fluorocarbons, chlorofluorocarbons and bromofluorocarbons are not flammable: they do not have carbon–hydrogen bonds to react and released halides quench flames.[120]
When all hydrogens are replaced with fluorine to achieve perfluoroalkanes, a great difference is revealed. Such compounds are extremely stable, and only sodium in liquid ammonia attacks them at standard conditions. They are also very insoluble, with few organic solvents capable of dissolving them.[119]
However, if a perfluorocarbon contains double or triple bonds (perfluoroalkenes or -alkynes), a very reactive towards ligand accepting result, even less stable than corresponding hydrocarbons.[121] Difluoroacetylene, which decomposes even under liquid nitrogen temperatures,[122] is a notable example. If such a molecule is asymmetric, then the more fluorinated carbon is attacked, as it holds positive charge caused by the C–F bonds and is shielded weakly[121] (similarly to that how unsaturated hydrocarbons attacked by HF add hydrogen to the more hydrogen-rich atom per Markovnikov's rule[123]).
Perfluorinated compounds, as opposed to perfluorocarbons, is the term used for molecules that would be perfluorocarbons—only carbon and fluorine atoms—except for having an extra functional group (even though another definition exists[124]). They share most of perfluorocarbon properties (inertness, stability, non-wettingness and insolubility in water and oils, slipperiness, etc.),[125] but may differ because of the functional group properties, although the perfluorocarbon tail differ the group-specific properties as compared to those of hydrocarbon-tailed compounds.
The perfluoroalkanesulfonic acids are also very notable for their acidity. The sulfonic acid derivative, trifluoromethanesulfonic acid, is comparable in strength to perchloric acid.[126] These compounds lower surface energy; for this reason, they, especially perfluorooctanesulfonic acid (PFOS, formerly the active component in brand "Scotchgard") have found industrial use as surfactants (see above).[126]
If a perfluorinated compound has a fluorinated tail, but also a few non-fluorinated carbons (typically two) near the functional group, it is called a fluorotelomer (such molecules are commercially treated as perfluorinated), but such molecules are more of industrial value than chemical. The chain end may similarly be attached to different functiuonal groups (via the hydrogenized terminal carbon), such as hydroxyl resulting in fluorotelomer alcohols, sulfonate resulting in fluorotelomer sulfonates, etc.[125]
7.2. Polymers
Fluoropolymers are similar in many regards with smaller molecules; adding fluorine to a polymer affects the properties in the same manner as in small molecules (increasing chemical stability, melting point, reducing flammability, solubility, etc.). Each fluoropolymer has own characteristic properties, though.[117]
The simplest fluoroplastic is polytetrafluoroethylene (PTFE, DuPont brand Teflon), which is a simple linear chain polymer with the repeating structural unit:–CF2–. PTFE has a backbone of carbons single bonded in a long chain, with all side bonds to fluorines. It contains no hydrogens and can be thought of as the perfluoro analog of polyethylene (structural unit: –CH2–). PTFE has high chemical and thermal stability, as expected for a perfluorocarbon, much stronger than polyethylene. Its resistance to van der Waals forces makes PTFE the only known surface to which a gecko cannot stick.[127] The compound, however, lacks an ability to transform upon melting, which is not a problem for various PTFE deratives, namely FEP (fluorinated ethylene propylene, with some fluorines replaced with the–CF3 group) or PFA (perfluoroalkoxy, some fluorines replaced with –OCF3). They share most properties with PTFE, but there are still differences, namely maximum usage temperature (highest for the non-flexible PTFE).[128]
There are other fluoroplastics other than perfluorinated. Polyvinylidene fluoride (PVDF, structural unit: –CF2CH2–), is an analog of PTFE with half the fluorines. PVF (polyvinyl fluoride, structural unit: –CH2CHF–) contains one one-fourth the fluorines of PTFE. Despite this, it still has many properties of more fluorinated compounds.[129] PCTFE (polychlorotrifluoroethylene, structural unit: –CF2CFCl–) is another important compound. It differs from PTFE by having a quarter of fluorine replaced with chlorines, yet this difference brings even greater hardness, creep resistance, and moisture persistence.[129]
Mild fluorination of polyethylene gives does not make all of the plastic lose its hydrogens for fluorine; only a thin layer (0.01 mm at maximum) is then affected. This is somewhat similar to metal passivation: the bulk properties are not affected, but the surface properties are, most notably, a greater vapor barrier. Therefore, they are a cheaper alternative to the perfluoro plastics if only surface is important.[130]

Nafion is a structurally complicated polymer. It has a PTFE-like backbone, but also contains side chains of perfluoro ether that end in sulfonic acid(–SO2OH) groups. It also possesses great chemical stability, while exact properties vary with morphology. However, because of the difficult chemical structure, it is also relatively easily converted to an ionomer (shows conductivity) by adding cations like Na+ or by converting into the sulfonic acid rather than the given sulfonyl fluoride. The conductivity is due to that the main carbon chain separates from the side chains, thus forming polar and non-polar regions. This form is also very hydroscopic.[131]
Fluoroelastomers, like other elastomers (artificial rubbers), consist of disordered polymer chains connected in three dimensions. The main challenges in making fluorelastomers are cross-linking (reacting the unreactive polymers), as well as removing the HF formed during curing. There are three main families of fluoroelasters. VDF/HFP is a copolymer system of vinylidene fluoride and (at least 20%) hexafluoropropylene. TFE/propylene is another copylymer system with better chemical resistance to some solvents. TFE/PMVE (perfluoromethylvinyl ether) is a copolymer system which creates a perfluorinated fluoroelastomer.[132]
The content is sourced from: https://handwiki.org/wiki/Chemistry:Compounds_of_fluorine
References
- Cheng, H.; Fowler, D. E.; Henderson, P. B.; Hobbs, J. P.; Pascaloni, M. R. (1999). "On the magnetic susceptibility of fluorine". Journal of Physical Chemistry A 103 (15): 2861–2866. doi:10.1021/jp9844720. Bibcode: 1999JPCA..103.2861C. https://dx.doi.org/10.1021%2Fjp9844720
- Mackay, Mackay & Henderson 2002, p. 72.
- Greenwood & Earnshaw 1998, p. 804.
- Macomber, Roger S. (1996). Organic chemistry. 1. University Science Books. p. 230. ISBN 978-0-935702-90-3. https://books.google.com/books?id=HyuogOtzoaYC&pg=PA230. Retrieved 26 July 2011.
- Hemat, R. A. S. (2009). Water. 42. Urotext. pp. 707–709. ISBN 9781903737125.
- Greenwood & Earnshaw 1998, p. 801.
- Akhmetov, N. S. (2001) (in ru) (4th ed.). Академик [Akademik]. p. 317. ISBN 978-5-06-003363-2.
- Jaccaud et al. 2005, p. 2.
- Nelson, Eugene W. (1947). "'Bad man' of the elements". Popular Mechanics 88 (2): 106–108, 260. https://books.google.com/books?id=1t8DAAAAMBAJ&pg=PA106.
- Lidin, Molochko & Andreeva 2000, pp. 442–455.
- Greenwood & Earnshaw 1998, p. 844.
- Jaccaud et al. 2005, p. 3.
- Pitzer, Kenneth S. (1975). "Fluorides of radon and element 118". Journal of the Chemical Society, Chemical Communications (18): 760b–761. doi:10.1039/C3975000760B. https://escholarship.org/uc/item/8xz4g1ff.
- Khriachtchev, Leonid; Pettersson, Mika; Runeberg, Nino; Lundell, Jan; Räsänen, Markku (2000). "A stable argon compound". Nature 406 (6798): 874–876. doi:10.1038/35022551. PMID 10972285. Bibcode: 2000Natur.406..874K. https://dx.doi.org/10.1038%2F35022551
- Lidin, Molochko & Andreeva 2000, p. 252.
- Moore, John W.; Stanitski, Conrad L.; Jurs, Peter C. (2010). Principles of chemistry: The molecular Science. Cengage Learning. p. 156. ISBN 978-0-495-39079-4. https://books.google.com/books?id=ZOm8L9oCwLMC&pg=PA156. Retrieved 7 May 2011.
- Wiberg, Wiberg & Holleman 2001, p. 422.
- Schlöder, T.; Riedel, S. (2012). "Investigation of Heterodimeric and Homodimeric Radical Cations of the Series: [F2O2]+, [F2Cl2]+, [Cl2O2]+, [F4]+, and [Cl4]+". RSC Advances 2 (3): 876–881. doi:10.1039/C1RA00804H. https://dx.doi.org/10.1039%2FC1RA00804H
- Young, J. P.; Haire, R. G.; Peterson, J. R.; Ensor, D. D.; Fellow, R. L. (1981). "Chemical consequences of radioactive decay. 2. spectrophotometric study of the ingrowth of berkelium-249 and californium-249 into halides of einsteinium-253". Inorganic Chemistry 20 (11): 3979–3983. doi:10.1021/ic50225a076. https://dx.doi.org/10.1021%2Fic50225a076
- Hulet, E. K. (1967). "Mendelevium: Divalency and Other Chemical Properties". Science 158 (3800): 486–8. doi:10.1126/science.158.3800.486. PMID 17749069. Bibcode: 1967Sci...158..486H. https://dx.doi.org/10.1126%2Fscience.158.3800.486
- Kratz, J. V. (2003). "Critical evaluation of the chemical properties of the transactinide elements (IUPAC Technical Report)". Pure and Applied Chemistry 75 (1): 103. doi:10.1351/pac200375010103. https://dx.doi.org/10.1351%2Fpac200375010103
- Kahn, Bernd (2007). Radioanalytical chemistry. ISBN 978-0-387-34122-4. https://books.google.com/books?id=9G8fsaXOH9AC&pg=PA356. Retrieved 4 May 2011.
- Lower numbers mean greater acidity; note the scale is logarithmical.
- Siegemund et al. 2005, p. 29.
- "Acids and bases". askthenerd.com. 2011. http://www.askthenerd.com/ocol/STRUCT/SB/17.HTM. Retrieved 18 August 2012.
- Olah, George A. (2005). "Crossing conventional boundaries in half a century of research". Journal of Organic Chemistry 70 (7): 2413–2429. doi:10.1021/jo040285o. PMID 15787527. https://dx.doi.org/10.1021%2Fjo040285o
- Hwang, I. C.; Seppelt, K. (2001). "Gold Pentafluoride: Structure and Fluoride Ion Affinity This work was supported by the Deutsche Forschungsgemeinschaft and the Fond der Chemischen Industrie". Angewandte Chemie International Edition 40 (19): 3690–3693. doi:10.1002/1521-3773(20011001)40:19<3690::AID-ANIE3690>3.0.CO;2-5. PMID 11592224. https://dx.doi.org/10.1002%2F1521-3773%2820011001%2940%3A19%3C3690%3A%3AAID-ANIE3690%3E3.0.CO%3B2-5
- Pauling, Linus A. (1960). The nature of the chemical bond and the structure of molecules and crystals: An introduction to modern structural chemistry. Cornell University Press. pp. 454–464. ISBN 978-0-8014-0333-0. https://archive.org/details/natureofchemical0000paul_3ed.
- Atkins, Peter; Jones, Loretta (2008). Chemical principles: The quest for insight. W. H. Freeman & Co. pp. 184–185. ISBN 978-1-4292-0965-6. https://books.google.com/books?id=4R6hb1OIMRUC&pg=PA184.
- Emsley, John (1981). "The hidden strength of hydrogen". New Scientist 91 (1264): 291–292. https://books.google.com/books?id=ZbthaZCUXy4C&pg=PA292. Retrieved 25 December 2012.
- Greenwood & Earnshaw 1998, pp. 812–816.
- Wiberg, Wiberg & Holleman 2001, p. 425.
- See citation for more detailed explanation.[36]
- Chambers, C.; Holliday, A. K. (1975). Modern inorganic chemistry (An intermediate text). The Butterworth Group. pp. 328–329. http://files.rushim.ru/books/neorganika/Chambers.pdf. Retrieved 2013-05-03.
- Hannan, Henry J. (1975). Technician's formulation handbook for industrial and household cleaning Products. Lulu.com. p. 31. ISBN 9780615156019. https://books.google.com/books?id=s3Ru9fvsGh4C&pg=PA31.
- Hannan, Henry J. (2010). Course in chemistry for IIT-JEE 2011. Tata McGraw Hill Education Private Limited. pp. 15–22. ISBN 9780070703360. https://books.google.com/books?id=wNYMUeCcaSEC&pg=SA15-PA22.
- Greenwood & Earnshaw 1998, pp. 816–819.
- Greenwood & Earnshaw 1998, p. 819.
- Lide 2004, p. 4-76.
- Lide 2004, p. 4-71.
- Lide 2004, p. 4-92.
- Oxtoby, David W.; Gillis, H. Pat; Campion, Alan (2012). Principle of Modern Chemistry. Cengage Learning. p. 693. ISBN 9780840049315.
- null
- Storer, Frank Humphreys (1864). First Outlines of a Dictionary of Solubilities of Chemical Substances. Cambridge. pp. 278–80. ISBN 978-1-176-62256-2.
- Remy, Heinrich (1956). Treatise on Inorganic Chemistry: Introduction and main groups of the periodic table. Elsevier Publishing Company. p. 383.
- Emeléus & Sharpe 1983, pp. 256–277.
- Walsh, Kenneth A. (2009-01-01). Beryllium chemistry and processing. ASM International. pp. 99–102, 118–119. ISBN 978-0-87170-721-5. https://books.google.com/books?id=3-GbhmSfyeYC&pg=PA119&dq=beryllium+fluoride+covalent#v=onepage.
- Mackay, Mackay & Henderson 2002, p. 243–244.
- Hertz, Raymond K. (1987). "General analytical chemistry of beryllium". in Coyle, Francis T.. Chemical analysis of metals: a symposium. ASTM. pp. 74–75. ISBN 978-0-8031-0942-1. https://books.google.com/books?id=uaWTfwrG644C&pg=PA74&dq=beryllium+fluoride+covalent#v=onepage.
- F. W. B. Einstein; P. R. Rao; James Trotter; Neil Bartlett (1967). "The crystal structure of gold trifluoride". Journal of the Chemical Society A: Inorganic, Physical, Theoretical 4: 478–482. doi:10.1039/J19670000478. https://dx.doi.org/10.1039%2FJ19670000478
- Wiberg, Wiberg & Holleman 2001, p. 1286.
- Wiberg, Wiberg & Holleman 2001, p. 1047.
- Sobolev, Boris Petrovich (2001). The Rare Earth Trifluorides: Introduction to materials science of multicomponent metal fluoride crystals. Institut d'Estudis Catalans. p. 51. ISBN 978-84-7283-610-5.
- Kern, S.; Hayward, J.; Roberts, S.; Richardson, J. W.; Rotella, F. J.; Soderholm, L.; Cort, B.; Tinkle, M. et al. (1994). "Temperature Variation of the Structural Parameters in Actinide Tetrafluorides". The Journal of Chemical Physics 101 (11): 9333–9337. doi:10.1063/1.467963. Bibcode: 1994JChPh.101.9333K. https://zenodo.org/record/1232099.
- Brown, Paul L.; Mompean, Federico J.; Perrone, Jane; Illemassène, Myriam (2005). Chemical Thermodynamics of Zirconium. Gulf Professional Publishing. p. 144. ISBN 978-0-444-51803-3. https://books.google.com/books?id=DvqwTdVhjMEC&pg=PA144.
- Lide 2004, pp. 4-60, -76, -92, -96.
- Becker, S.; Müller, B. G. (1990). "Vanadium Tetrafluoride". Angewandte Chemie International Edition in English 29 (4): 406–407. doi:10.1002/anie.199004061. https://dx.doi.org/10.1002%2Fanie.199004061
- Brown, David; Canterford, J. H.; Colton, Ray (2009). Halides of the Transition Elements: Halides of the first row transition metals, by R. Colton and J. H. Canterford. Wiley. p. 213. https://books.google.com/books?id=grdPAAAAYAAJ&q=%22manganese+tetrafluoride%22&dq=%22manganese+tetrafluoride%22. Retrieved 2012-06-06.
- Nakajimȧ, Tsuyoshi; Žemva, Boris; Tressaud, Alain (2009). Advanced Inorganic Fluorides: Synthesis, Characterization, and Applications (1st ed.). Elsevier. p. 111. ISBN 978-0444720023. https://archive.org/details/advancedinorgani00naka.
- Emeléus & Sharpe 1983, p. 105.
- Breunig, Hans Joachim. "Bismuth compounds". Kirk-Othmer Encyclopedia of Chemical Technology Volume 4. John Wiley & Sons. p. 22. https://www.scribd.com/doc/30120462/Bismuth-Compounds.
- Wiberg, Wiberg & Holleman 2001, p. 770.
- Suzuki, Hitomi; Matano, Yoshihiro (2001). Organobismuth chemistry. Elsevier. p. 8. ISBN 978-0-444-20528-5. https://archive.org/details/organobismuthche00suzu_245.
- Bartlett, Neil; Lohmann, D. H. (1962). "Dioxygenyl hexafluoroplatinate (V), O2+[PtF6]−". Proceedings of the Chemical Society (3): 115. doi:10.1039/PS9620000097. https://dx.doi.org/10.1039%2FPS9620000097
- Bartlett, Neil (1962). "Xenon hexafluoroplatinate (V) Xe+[PtF6]−". Proceedings of the Chemical Society (6): 218. doi:10.1039/PS9620000197. https://dx.doi.org/10.1039%2FPS9620000197
- Wiberg, Wiberg & Holleman 2001, p. 594.
- Vogt, T.; Fitch, A. N.; Cockcroft, J. K. (1994). "Crystal and molecular structures of rhenium heptafluoride". Science 263 (5151): 1265–67. doi:10.1126/science.263.5151.1265. PMID 17817431. Bibcode: 1994Sci...263.1265V. https://dx.doi.org/10.1126%2Fscience.263.5151.1265
- Bayerische Julius-Maximilians-Universität Würzburg 2006, p. 93.
- Technische Universität Berlin (2012). "Prediction of new compounds and new oxidation states". http://www.quantenchemie.tu-berlin.de/menue/research/applications/prediction_of_new_compounds_and_new_oxidation_states/. Retrieved 2012-05-24.
- Riedel, S.; Kaupp, M. (2009). "The highest oxidation states of the transition metal elements". Coordination Chemistry Reviews 253 (5–6): 606–624. doi:10.1016/j.ccr.2008.07.014. https://dx.doi.org/10.1016%2Fj.ccr.2008.07.014
- Bayerische Julius-Maximilians-Universität Würzburg 2006, p. 102.
- Weinstock, Bernard; Malm, John G. (September 1958). "Osmium Hexafluoride and its Identity with the Previously Reported Octafluoride". Journal of the American Chemical Society 80 (17): 4466–4468. doi:10.1021/ja01550a007. https://dx.doi.org/10.1021%2Fja01550a007
- Weldkamp, Achim; Frenking, Gernot (1993). "Quantum-mechanical ab initio investigation of the transition-metal compounds OsO4, OsO3F2, OsO2F4, OsOF6, and OsF8". Chemische Berichte 126 (6): 1325–1330. doi:10.1002/cber.19931260609. https://dx.doi.org/10.1002%2Fcber.19931260609
- Noury, Stephane; Silvi, Bernard; Gillespie, Ronald J. (2002). "Chemical bonding in hypervalent molecules: Is the octet rule relevant?". Inorganic Chemistry 41 (8): 2164–2172. doi:10.1021/ic011003v. PMID 11952370. http://alpha.chem.umb.edu/chemistry/Seminar/06-09%20WQE/InorgI.Carter.pdf. Retrieved 23 May 2012.
- Greenwood & Earnshaw 1998, pp. 198–199.
- Martinie, R. J.; Bultema, J. J.; van der Wal, M. N.; Burkhart, B. J.; van der Griend, D. A.; de Kock, R. L. (2011). "Bond order and chemical properties of BF, CO, and N2". Journal of Chemical Education 88 (8): 1094–1097. doi:10.1021/ed100758t. Bibcode: 2011JChEd..88.1094M. http://www.calvin.edu/~dav4/Documents/ed100758t.pdf.
- Ellis, Bryan David (2001). Scientific essentialism. Cambridge University Press. p. 69. ISBN 978-0521800945.
- Aigueperse et al. 2005, p. 28.
- Aigueperse et al. 2005, p. 30.
- Raghavan, P. S. (1998). Concepts and problems in inorganic Chemistry. Discovery Publishing House. pp. 164–165. ISBN 9788171414185. https://books.google.com/books?id=pBiS0jc-kWIC&pg=PA164.
- Aigueperse et al. 2005, p. 37.
- Norman, Nicholas C. (1998). Chemistry of arsenic, antimony and bismuth. Springer. p. 97. ISBN 978-0751403893. https://books.google.com/books?id=vVhpurkfeN4C&pg=PA98.
- Christe, K. O.; Wilson, W. W. (1986). "Synthesis and characterization of NF+4BrF−4 and NF+4BrF4O−". Inorganic Chemistry 25 (11): 1904–1906. doi:10.1021/ic00231a038. https://dx.doi.org/10.1021%2Fic00231a038
- Harbison, G. S. (2002). "The Electric Dipole Polarity of the Ground and Low-lying Metastable Excited States of NF". Journal of the American Chemical Society 124 (3): 366–367. doi:10.1021/ja0159261. PMID 11792193. https://dx.doi.org/10.1021%2Fja0159261
- Davis, Steven J.; Rawlins, Wilson T.; Piper, Lawrence G. (1989). "Rate coefficient for the H + NF(a1Δ) reaction". The Journal of Physical Chemistry 93 (3): 1078–1082. doi:10.1021/j100340a013. http://metastablestates.com/Publications/JPC_93_1078_1989.pdf.
- Murthy, C. Parameshwara (2008). University chemistry, Tom 1. New Age International. pp. 180–182, 206–208. ISBN 978-8122407426.
- Crawford, M.; Klapötke, T. M. (1999). "The trifluorooxonium cation, OF+3". Journal of Fluorine Chemistry 99 (2): 151–156. doi:10.1016/S0022-1139(99)00139-6. https://dx.doi.org/10.1016%2FS0022-1139%2899%2900139-6
- Greenwood & Earnshaw 1998, p. 639.
- Greenwood & Earnshaw 1998, p. 686.
- Wiberg, Wiberg & Holleman 2001, p. 435.
- Wiberg, Wiberg & Holleman 2001, p. 436.
- Pitzer, Kenneth Sanborn, ed (1993). Molecular structure and statistical thermodynamics: Selected papers of Kenneth S. Pitzer. 1. World Scientific. p. 111. ISBN 978-9810214395.
- Gmelin, Leopold (1985-07-03). Gmelin handbook of inorganic chemistry: At—Astatine (8th ed.). Springer-Verlag. p. 224. ISBN 9783540935162.
- Greenwood & Earnshaw 1998, pp. 828–830.
- Patnaik, Pradyot (2007). A comprehensive guide to the hazardous properties of chemical substances. John Wiley & Sons. pp. 478–479. ISBN 9780471714583. https://books.google.com/books?id=-CRRJBVv5d0C&pg=PA478.
- "The Nobel Prize in chemistry 1994". nobelprize.org. http://nobelprize.org/nobel_prizes/chemistry/laureates/1994/index.html. Retrieved 22 December 2008.
- Wiberg, Wiberg & Holleman 2001, pp. 392–393.
- Chemical and Engineering News as cited by Michael Barnes. "Neil Bartlett, emeritus professor of chemistry, dies at 75". University of California Newsroom. http://www.universityofcalifornia.edu/news/article/18376. Retrieved 24 December 2011.
- Wiberg, Wiberg & Holleman 2001, p. 438.
- Wiberg, Wiberg & Holleman 2001, p. 400.
- Grosse, A. V.; Kirshenbaum, A. D.; Streng, A. G.; Streng, L. V. (1963). "Krypton Tetrafluoride: Preparation and Some Properties". Science 139 (3559): 1047–1048. doi:10.1126/science.139.3559.1047. PMID 17812982. Bibcode: 1963Sci...139.1047G. https://dx.doi.org/10.1126%2Fscience.139.3559.1047
- Dixon, D. A.; Wang, T. H.; Grant, D. J.; Peterson, K. A.; Christe, K. O.; Schrobilgen, G. J. (2007). "Heats of Formation of Krypton Fluorides and Stability Predictions for KrF4 and KrF6 from High Level Electronic Structure Calculations". Inorganic Chemistry 46 (23): 10016–10021. doi:10.1021/ic701313h. PMID 17941630. https://dx.doi.org/10.1021%2Fic701313h
- There is no general line where oxidation states are "relatively low" or "relatively high", they rely on specific elements (and defined only for elements that have highest oxides and fluorides are in different oxidation states); in general, +7 and +8 are high, while +4 and below are low. States +5 and +6 rely on element properties, like atomic radius; for a small nitrogen atom, +5 is "high" here, but for larger palladium and platinum +6 is still "low".
- Wiberg, Wiberg & Holleman 2001, p. 398.
- Riedel, S.; Kaupp, M. (2009). "The highest oxidation states of the transition metal elements". Coordination Chemistry Reviews 253 (5–6): 606–624. doi:10.1016/j.ccr.2008.07.014. https://dx.doi.org/10.1016%2Fj.ccr.2008.07.014
- "Technical data sheet 320 Ruthenium tetroxide 0.5% aqueous solution". Polysciences, Inc. 2000. p. 1. http://www.polysciences.com/SiteData/poly/Assets/DataSheets/320.pdf. Retrieved 28 December 2012.
- Bayerische Julius-Maximilians-Universität Würzburg 2006, p. 34.
- Note that aside from the molecular one, other forms of the highest fluorides are not considered to be possible. Such compounds are extremely unlikely to be completely ionic because of large sixth, seventh, and eighth oxidation energies of all elements, which make the reactions that may produce such compounds highly unfavorable (even though the possible osmium octafluoride may be mostly ionic[76]).
- Jensen, William B. (2008). "Is mercury now a transition element?". Journal of Chemical Education 85 (9): 1182–1183. doi:10.1021/ed085p1182. Bibcode: 2008JChEd..85.1182J. https://dx.doi.org/10.1021%2Fed085p1182
- Wang, Xuefang; Andrews, Lester; Riedel, Sebastian; Kaupp, Martin (2007). "Mercury is a transition metal: The first experimental evidence for HgF4". Angewandte Chemie 119 (44): 8523–8527. doi:10.1002/ange.200703710. https://dx.doi.org/10.1002%2Fange.200703710
- Lewars 2008, pp. 60–61.
- O'Hagan, D. (2008). "Understanding organofluorine chemistry. An introduction to the C–F bond". Chemical Society Reviews 37 (2): 308–319. doi:10.1039/b711844a. PMID 18197347. https://dx.doi.org/10.1039%2Fb711844a
- Siegemund et al. 2005, p. 2.
- Siegemund et al. 2005, p. 4.
- Jaccaud et al. 2005, p. 13.
- Siegemund et al. 2005, pp. 3–5.
- Carlson & Scmiegel 2005, p. 3.
- Okazoe, Takashi (2009). "Overview on the history of organofluorine chemistry from the viewpoint of material industry" (PDF). Proceedings of the Japan Academy, Series B 85 (8): 276–289. doi:10.2183/pjab.85.276. PMID 19838009. PMC 3621566. Bibcode: 2009PJAB...85..276O. https://www.jstage.jst.go.jp/article/pjab/85/8/85_8_276/_pdf.
- Siegemund et al. 2005, pp. 7–8.
- Sukornick, B. (1989). "Potentially acceptable substitutes for the chlorofluorocarbons". International Journal of Thermophysics 10 (3): 553–561. doi:10.1007/BF00507978. Bibcode: 1989IJT....10..553S. https://dx.doi.org/10.1007%2FBF00507978
- Siegemund et al. 2005, p. 18.
- Urben, Peter (2006). Bretherick's Handbook of Reactive Chemical Hazards (7th ed.). Surendra Kumar. p. 269. ISBN 9780123725639.
- Siegemund et al. 2005, p. 5.
- Mendicino, L. (1999). Environmental issues in the electronics and semiconductor industries. The Electrochemical Society. p. 116. ISBN 9781566772303.
- Knepper, Thomas P.; Lange, Frank T. (2011). Polyfluorinated Chemicals and Transformation Products. Springer. p. 27. ISBN 9783642218712. https://archive.org/details/polyfluorinatedc00knep.
- Siegemund et al. 2005, p. 32.
- University of California, Berkeley. "Research into gecko adhesion". Archived from the original on 14 October 2007. https://web.archive.org/web/20071014063923/http://socrates.berkeley.edu/~peattiea/research_main.html. Retrieved 29 April 2011.
- Carlson & Scmiegel 2005, pp. 3–4.
- Carlson & Scmiegel 2005, p. 4.
- Kharitonov, A. P.; Taege, R.; Ferrier, G.; Piven, N. P. (2005). "The kinetics and mechanism of the direct fluorination of polyethylenes". Surface Coatings International Part B: Coatings Transactions 88 (3): 201–212. doi:10.1007/BF02699574. http://www.airproducts.de/fluorination/fluortechnologie/pdf/kinetics-mechanism-fluorination-polyethylenes.pdf. Retrieved 2013-05-03.
- Rhoades, David Walter (2008). Broadband dielectric spectroscopy studies of Nafion. p. 2. ISBN 9780549785408.
- Martin, John Wilson (2007). Concise encyclopedia of the structure of materials. Elsevier. pp. 187–194. ISBN 978-0-08-045127-5. https://books.google.com/books?id=xv420pEC2qMC&pg=PA189.