Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Several reports have shown that the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has the potential to also be neurotropic. Four Adverse Outcome Pathways (AOPs) leading to neurological adverse outcomes (AO), anosmia, encephalitis, stroke, and seizure, were developed. Biological key events (KEs) identified to induce these AOs included binding to ACE2, blood–brain barrier (BBB) disruption, hypoxia, neuroinflammation, and oxidative stress. The modularity of AOPs allows the construction of AOP networks to visualize core pathways and recognize neuroinflammation and BBB disruption as shared mechanisms.
- AOP
- SARS-CoV-2
- neuropathology
- anosmia
1. ACE2 Receptor Interaction
A classical Adverse Outcome Pathways (AOPs) generally starts with an molecular initiating event (MIE). In the present case, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was identified as the initiating stressor. Several studies have shown that SARS-CoV-2 enters cells by binding the surface S-protein to the angiotensin-converting enzyme 2 (ACE2) cell-surface receptor (Event 1739 in AOP Wiki) [1][2][3][4]. After the initial binding, transmembrane protease serine 2 (TMPRSS2), which is anchored to the outer surface of the host cell membrane, has been shown to cleave the S-protein at a specific site, facilitating a conformational change in the latter that promotes virus-cell membrane fusion and subsequent viral entry into the host cell [2][5]. The ACE2 receptor and TMPRSS2 are expressed in a wide variety of tissues, including regions of the brain. A recent study identified the expression levels of ACE2 in both endothelial cells and non-vascular cells, composed predominantly of neurons but also with low expressions of astrocytes and microglia [6]. The expression varied among the brain regions with high levels in the pons, visual cortex, and amygdala, and reduced levels in the midbrain, cerebellar cortex, dentate nucleus, and medulla. However, it is important to note that not all the cells of the nervous system possess the machinery that is necessary for the interaction with SARS-CoV-2 [7], and it is possible that the virus may infect certain cells of neuronal origin through a different set of extracellular host receptors, such as neuropilin-1 [8]. In this respect, neuropilin-1 has been detected in olfactory epithelium ensheathing cells and there is some evidence showing that SARS-CoV-2 can enter the brain through the olfactory nerve [9][10]. It has also been suggested that SARS-CoV-2 S-protein can produce an inflammatory response in brain endothelial cells, resulting in the disruption of the blood–brain barrier (BBB) integrity and enabling the virus to enter the brain [11]. Furthermore, it has been shown that the S-protein can impair the vascular and immune regulatory functions of brain pericytes, which could explain vascular-mediated brain damage [12]. However, to date, there has been limited detection of viral proteins in the brain and cerebrospinal fluid (CSF) [13][14], indicating that the neurological effects may be secondary due to damage to endothelial cells and the trafficking of cytokines into the brain from the blood [15]. Concordantly, recent publications rule out the possibility that the axons of olfactory sensory neurons (OSNs) constitute a virus gateway to the central nervous system (CNS) [16][17], but the transsynaptic transfer of SARS-CoV-2 from peripheral neuron infection remains a possibility. Consequently, the MIEs and earlier key events (KEs) leading to adverse outcomes (AOs) in the nervous system need to be further evaluated and confirmed.
From a multiscale perspective, it is interesting to consider that the focus should not only be on ACE2 as an initiating event in AOPs. Including environmental, social, and individual scales could help to mitigate the spread and prevent future pandemics. In addition, the precise SARS-CoV-2 molecular pathways might not be fully understood and may be altered depending on the target cell type and variants. Recent research has identified important non-ACE2 factors in excessive inflammation involving key monocytes and macrophages lacking ACE2 receptors pathways [18].
2. Blood–Brain Barrier (BBB) Disruption
The BBB is crucial in protecting the hemodynamic function of the brain. In addition, it is a key structural defense against toxic substances and microorganisms such as viruses and chemicals. The complex structure of the BBB, which comprises a number of cell types and junctions, mechanically and biochemically regulates the permeability of xenobiotics [19]. It is, therefore, not surprising to see the clinical reports of neuroinflammatory and BBB disruption markers described repeatedly in the literature. However, it remains an open question as to how and when these processes may occur. The interconnected nature of brain capillary endothelial cells (BCECs), pericytes, neurons, astrocytes, and microglia in the BBB, strongly suggest this to be a path of SARS-CoV-2 viral entry to the brain, and a contribution to neuroinflammatory events [20]. A compromised BBB is one of the proposed KEs in the network of neuro AOPs (Figure 1) linked to several AOs such as strokes, seizures, anosmia, and encephalitis [21]. Evidence from in vitro models showed that isolated spike proteins can cross the BBB [11][22]. Viral particles were also identified in the frontal lobe by Paniz-Mondolfi and colleagues, suggesting a hematological route of infection through the BBB [23]. ACE2 expression has been reported in the BBB [13][22], and although in low abundance, its blockage with anti-ACE2 antibody in human induced pluripotent stem cells (hiPSC) led to the almost complete blockage of infection [22]. Elevated expressions of TMPRSS2 and NRP1 were reported in an hiPS-derived BCEC model, and their role in SARS-CoV-2 infection was demonstrated with the use of specific antibodies and inhibitors [22]. Alterations of the interferon-γ-mediated signaling pathways in COVID-19 patients and in a cellular model [22] revealed another potential explanation for the compromised BBB, specifically through the disruption of the brain endothelial cells [24] resulting in increased proinflammatory cytokines within an individual even without viral entry into the brain.
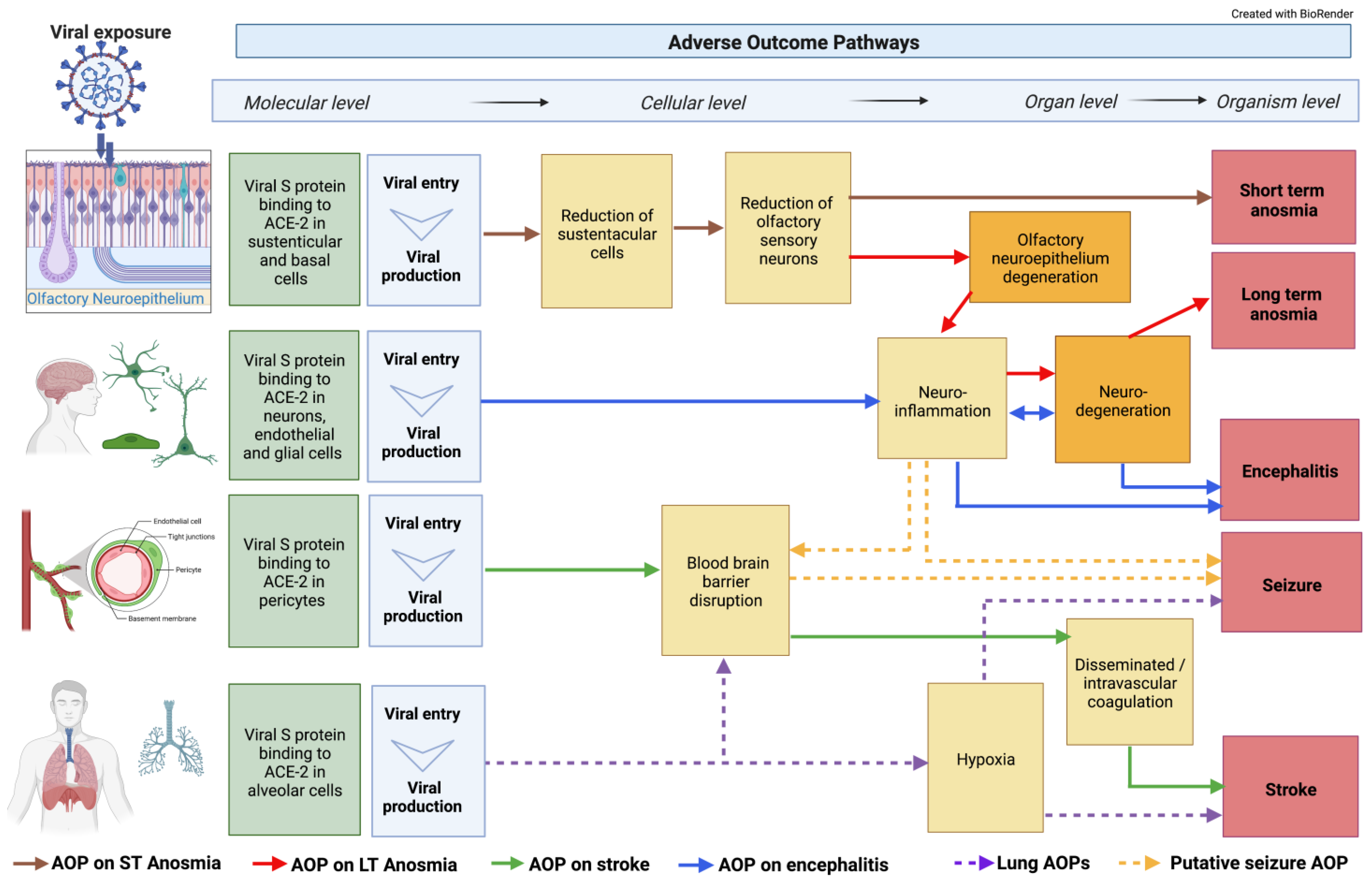
Figure 1. Integration of classical AOPs. Neuronal-related AOPs developed so far within the CIAO dedicated neuro working group integrated into an AOP network. Some of these AOPs are already uploaded on the AOP-Wiki, whereas others are only drafted outside the platform. The AOP network was built on 18 KEs, including 4 MIEs, 8 specialized (neuro-related) Kes, and 4 AOs. Two of these KEs were already available in the AOP-Wiki while the others were developed within the CIAO project. The dotted lines indicate not fully developed AOPs.
3. Hypoxia and Hypoxemia
Hypoxia and hypoxemia are KEs involved in severe neurological complications seen in COVID-19. Brain injury from hypoxia and hypoxemia are commonly reported in fatal COVID-19. Two separate studies showed that all enrolled patients who died had acute hypoxic damage, without evidence of thrombi or vasculitis, throughout many brain structures after infection with SARS-CoV-2 [25][26]. Frontera and colleagues identified hypoxemia, along with sepsis and uremia to be among the most common causes of multifactorial toxic-metabolic encephalopathy (TME) [27]. Of these factors, hypoxia was the risk factor most associated with TME, thus supporting the hypothesis that hypoxia is one of the crucial key events [28][29]. However, hypoxia is also identified as a potential mechanism in stroke (see below). In Figure 1, hypoxia is identified as a KE in lung, stroke, and seizure AOPs; however, the KER is still under development.
4. Neuroinflammation and Oxidative Stress
Neuroinflammation is a crucial event in the onset and consolidation of SARS-CoV-2-related neurological sequelae and neurodegenerative illnesses [30][31]. Neuroinflammation can be detected by the presence of activated microglia and astrocytes. Patients with moderate to severe COVID-19 are often characterized by the presence of elevated plasma levels of glial fibrillary acidic protein (GFAP), which is a biochemical indicator of astrocytic activation [32]. Changes in glial cell morphology, increased antigen expression, and the increased proliferation of the glial cells in the affected brain regions represent classical hallmarks of neuroinflammation [33][34][35][36][37][38].
Both astrocytes and microglia modulate the activation of inflammatory signaling pathways. After this, there is increased expression and/or release of inflammatory cytokines’, eicosanoids’, and metalloproteinases’ [39] production of reactive oxygen (ROS) and nitrogen species (RNS) [40]. Microglia and astrocytes vary in their activation and there is an interplay between pro-inflammatory, anti-inflammatory signaling, and cellular functions, including phagocytosis [36][41]. Rather than a direct migration of the virus into the brain through the nasal cavity and the olfactory pathway, or penetration of the virus across the BBB, immune activation, the entering of activated immune cells due to disrupted BBB, and inflammation within the brain are currently considered as the most plausible triggers of neurologic disease occurring as a consequence of SARS-CoV-2 infection in acute COVID-19 [15]. As part of the BBB, astrocytes can receive circulating pro-inflammatory cytokine signals, and/or activate microglia leading to neuroinflammation [42]. Patients affected by SARS-CoV-2 can present with a large increase in systemic pro-inflammatory cytokines and the induction of reactive pro-inflammatory microglia, which can upregulate the expression of neuroinflammation-related genes [28][39][40]. Analysis of cerebrospinal fluid (CSF) samples from COVID-19 patients showed the upregulation of interferon-regulated gene expression in dendritic cells, the activation of T cells and NK cells, as well as elevated levels of IL1 and IL12 [43]. This immune response seems to be compartmentalized, as suggested by the presence of clonal expansion of T cells and antibodies specific for SARS-CoV-2 spike protein in the CSF [44]. Additionally, specific markers of monocyte activation and neuronal injury were also found in the CSF during this acute phase of the disease [45].
5. Neurodegeneration
Several pathological outcomes have been proposed and are under investigation to determine how SARS-CoV-2-induced damage is associated with neurodegeneration. SARS-CoV-2-induced systemic inflammation, or the cytokine storm, may cause disruption of the BBB, neural and glial cell damage or dysfunction, and penetration of pro-inflammatory cytokines into the CNS. These pro-inflammatory events could for example alter the ability of microglial cells to phagocytose amyloid beta, promoting the accumulation of amyloid plaques, a known hallmark of Alzheimer’s disease (AD) [46]. In addition, systemic inflammation could cause the activation of neural-immune cells for the further induction of pro-inflammatory cytokine production in the brain. Subsequently, these cytokines could contribute to synaptic dysfunction and neuronal loss and lead to neurodegeneration associated with, for example, AD, Parkinson’s disease, and multiple sclerosis [47].
SARS-CoV-2 induces hypoxic alterations and demyelinating lesions in the brains of some individuals [22][46][47]. The outcomes of these alterations have been shown in follow-up studies of recovered SARS-CoV-2 patients, indicating alterations in brain functional integrity, specifically in the hippocampus. The hippocampus is the memory house of the brain, and its atrophy has been linked to cognitive decline [48][49]. Furthermore, severely infected individuals have characteristic hypercoagulation and disseminated intravascular coagulation [50], which may lead to reduced perfusion of the white matter, potentially leading to ischemic white matter damage. SARS-CoV-2-induced hypoperfusion in the brain can also increase the phosphorylation of tau, the main component of neurofibrillary tangles found in AD-affected brains. This is a characteristic feature of the early stage of AD and cognitive decline [51][52]. Furthermore, it has been suggested that SARS-CoV-2-induced neurotropism may lead to an increased risk of AD development and the progression of AD-like symptoms [53]. SARS-CoV-2 infection in the CNS can lead to neuroinflammation, which activates downstream signaling, including the increased release of pro-inflammatory markers, intense oxidative stress, and ineffective innate immune responses [54][55][56]. In the presence of certain risk factors or comorbidities, neuroinflammation can become prolonged or uncontrollable and can lead to an increased risk of neurodegeneration and disease [57]. Moreover, SARS-CoV-2-infected individuals with comorbidities that display existing inflammation, for example, diabetes, atherosclerosis, or sub-clinical dementia, could potentially be more at risk of neurodegenerative disorders.
6. Anosmia
Olfactory disturbances were the most usual neurological symptoms of COVID-19 for the initial variants of SARS-CoV-2. Loss of smell (anosmia, if complete loss of smell) has been included as a common symptom of COVID-19 [58]. The prevalence, intensity, and duration of these olfactory symptoms vary from patient to patient and are affected by epigenetic and geographic modulating factors [59][60]. The causes of the differences are not known and are driven by variability in the human population but also by the variation of SARS-CoV-2 variants that are currently in circulation. The self-rated and objective smell evaluation by patients also contributes to a different prevalence, the scoring being more subjective.
The exact mechanism of anosmia is still unknown, but there are probably several factors that contribute to the loss of smell. Developing an AOP based on current evidence from the literature enabled to propose underlying mechanisms and to identify current knowledge gaps guiding further research [61]. SARS-CoV-2 initiates with the binding of the viral spike proteins to the ACE2 receptors. In the olfactory epithelium, ACE2 protein is located mainly in the sustentacular cells but is not expressed in the olfactory receptor neurons (ORN) as demonstrated by ACE2 immunohistochemical expression [62]. The damage to the sustentacular cells leads to the subsequent damage of the ORN with the complete or partial loss of the normal sense of smell. The rapid regeneration of sustentacular cells due to stem cell maturation correlates to the recovery of the sense of smell that is clinically observed in most COVID-19 cases. The time course of smell recovery in COVID-19 is around one week [10] but can also last substantially longer in some individuals (long-term anosmia). Measuring olfactory function revealed changes in smell perception that could last up to 15 months after symptom onset. In these cases, alterations at the CNS level, showed also in a reorganization of ORN nuclear architecture and a widespread downregulation of olfactory receptors [63], could be associated with neuroinflammation leading to neurodegeneration-inducing persistent symptoms [64].
The mechanisms leading to short-term anosmia involving the olfactory neuroepithelium have been detailed in AOP394, entitled “SARS-CoV-2 infection of olfactory epithelium leading to impaired olfactory function (short-term anosmia)” which can be found in the AOP-Wiki [65]. This AOP describes the current mechanistic understanding of the causative links between the binding of spike protein to ACE2 receptors on sustentacular cells (MIE), which leads to sustentacular cell depletion, olfactory sensory neuron decreases, and olfactory epithelium degeneration (KEs), resulting in anosmia (AO) (Figure 1) [66].
7. Encephalitis
It has been recently suggested that a transient viral infection of the brain may occur in the early phase of infection, and/or that viral antigens may be present in the brain at low concentrations [15]. Although infrequently detected, infected CNS cells do not present with clusters of surrounding inflammatory cells, which suggests that the presence of the virus in the CNS may not stimulate classic viral encephalitis [15].
However, some studies have reported the presence of SARS-CoV-2 in the CNS and CSF of patients with acute neurologic symptoms specific to encephalitis [67][68]. Encephalitis has been clinically observed in COVID-19 patients. It is characterized by acute onset, and the symptoms include headache, fever, vomiting, convulsions, and consciousness disorders [67][69]. In the ongoing pneumonia epidemic, viral encephalitis was confirmed by the presence of SARS-CoV-2 in the CSF of patients with COVID-19 [70]. Moreover, the presence of SARS-CoV-2 viral particles in the endothelial cells and the pericytes of brain capillaries and neurons [20][70][71], as well as in the glial cells (microglia and astrocytes) [71] was observed in the post-mortem examinations of SARS-CoV-2-infected patients.
The binding of spike protein to ACE2 receptors on endothelial cells, pericytes of brain capillaries, as well as microglia and astrocytes, triggers their activation, which eventually results in the formation and release of proinflammatory cytokines/chemokines, nitric oxide, prostaglandin E2, ROS, and RNS. As a consequence, these proinflammatory factors can trigger neuronal cell death by well-known mechanisms [72][73][74] contributing, together with brain neuroinflammation, to encephalitis.
Currently, an AOP entitled “Binding of SARS-CoV-2 spike protein to ACE 2 receptors expressed on brain cells (neuronal and non-neuronal) leads to neuroinflammation resulting in encephalitis” is under development [75]; this AOP describes the current mechanistic understanding of the causative links between the binding of spike protein to ACE2 receptors on brain cells (MIE), which leads to glia activation causing neuroinflammation and neurodegeneration (KE), resulting in encephalitis (AO) (Figure 1).
8. Stroke
Large vessel strokes have been reported in a number of cases in patients diagnosed with COVID-19 [57][76][77]. As cerebrovascular disease is not a common clinical finding in patients with SARS-CoV-2, many predisposition factors such as cardiovascular comorbidities, including hypertension (HTN) and diabetes mellitus (DM), were proposed to play a role. However, age might not be a critical risk factor as there are reports showing younger patients with occlusions of large vessels linked to SARS-CoV-2 infection [76]. Since the beginning of the pandemic, patients diagnosed with stroke attributed to SARS-CoV-2 infection were also found to have elevated disseminated intravascular coagulation (DIC) and increased numbers of venous thromboembolisms [77].
The exact mechanism of COVID-19-induced stroke is not well established, although evidence has emerged indicating that SARS-CoV-2 can directly cause hypercoagulopathy, arteritis, and vascular endothelial dysfunction, which can lead to ischemic stroke [78]. Hypoxia and the excessive secretion of inflammatory cytokines have also been proposed as potential mechanisms involved in large vessel strokes in COVID-19 complications [57][79]. However, there are several reports hypothesizing that cerebrovascular disease in COVID-19 patients might not be due to the direct viral mechanism but rather the result of other conditions that are present in patients with severe COVID-19 during hospitalization (e.g., heart failure, septic shock, coagulopathy, and acute cardiac injury), which are well established factors potentially predisposing patients to stroke [76][77][80]. SARS-CoV-2 is known to block ACE2 receptors, which are regulators of blood pressure. This can lead to the functional underexpression of ACE2, thereby increasing the risk of hemorrhagic stroke. In individuals with hypertension, who already experience decreased expressions of ACE2 and difficulties with controlling blood pressure, SARS-CoV-2 infection may be more likely to create neurological complications characterized as cerebrovascular hemorrhage [76][79].
Markers that have been associated with cerebrovascular disease in patients positive for SARS-CoV-2 include elevated d-dimer or fibrin degradation product levels, reduced platelet counts, and transient increases in serum inflammatory cytokines and antiphospholipid antibodies [57][77][80]. The access of SARS-CoV-2 to cerebral vasculature is thought to be achieved through the general circulation, most likely by breaching the BBB and affecting the parenchyma [57].
An AOP entitled “Binding of SARS-CoV-2 spike protein to ACE 2 receptors expressed on pericytes leads to disseminated intravascular coagulation resulting in cerebrovascular disease (stroke)” is under development in the AOP Wiki [80]; this AOP describes the current mechanistic understanding of the causative links between the binding of spike protein to ACE2 receptors on pericytes (MIE), which leads to BBB disruption, DIC, and thrombosis (KEs), and results in cerebrovascular disease (AO) (Figure 1).
9. Seizure and Epilepsy
The AOP on seizure and epilepsy linked to COVID-19 is still under development but not yet in the AOP Wiki. Seizures are defined as uncontrolled electrical activity disturbances in the brain and can lead to both motor and behavioral symptoms [81]. Experiencing more than two seizures within 24 h is typical of epilepsy. In larger COVID-19 data sets, epilepsy is reported in 0.5–4% of patients [82][83][84]. Potential KEs that can lead to seizure in these patients includes hypoxia [83], neuroinflammation [84], and a compromised BBB [85][86][87] (Figure 1). A recent systematic review identified 62 manuscripts that reported on patients with COVID-19 and seizures [88]. Many of the papers were case studies describing patients with new onsets of focal seizures, serial seizures, and status epilepticus. The interpretation by the authors in this study was that organ failure, metabolic derangements, drug–drug interactions, or brain damage were potential causes of seizures. Though very rare, there have been case studies describing other SARS-CoV infections of the brain linked to seizures [89][90], indicating that other KEs may be involved in this AOP.
10. Integration of Classical AOPs
The modularity of AOPs allows the self-assembling and construction of AOP networks. Through an AOP network, shared KEs and key event relationships (KERs) are emerging, and knowledge gaps are being identified [91][92]. Furthermore, the assembling of AOP networks provides an opportunity to better visualize the commonalities and core pathways and mechanisms (Figure 1). Through the AOP network, the viral spike protein binding to ACE2 located in endothelial, neuronal, and glial cells is presented as an important step for long-term anosmia compared to short-term anosmia, which involves binding to ACE2 found in the sustentacular and basal cells of the olfactory epithelium. Neuroinflammation is emerging as a common KE for many individual AOPs that describe the disease process that can lead to long-term anosmia, encephalitis, stroke, and seizures.
The recognition of the role of low brain oxygen also helps to elucidate the connection of neurological symptoms to the well-studied pulmonary distress identified with COVID-19. Hypoxemia and hypoxia serve as illustrations of how multiple systems, in this case, pulmonary and neuronal, can recursively interact and lead to AOs (Figure 1). Moreover, it is an example of how mechanisms ranging from the molecular to higher biological levels in COVID-19 pathways can be integrated to better understand the disease.
Within the CIAO project, there is a parallel effort to create and publish a more comprehensive AOP network that covers all the possible mechanisms and AOs induced by SARS-CoV-2 beyond the nervous system. This assembling of neuro-related AOPs into a network as described in Figure 1 was the outcome of the neuro working group discussion and is open to revision as new data become available and as potential new KEs and/or pathways emerge.
This entry is adapted from the peer-reviewed paper 10.3390/cells11213411
References
- Xu, X.; Chen, P.; Wang, J.; Feng, J.; Zhou, H.; Li, X.; Zhong, W.; Hao, P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020, 63, 457–460.
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280 e278.
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273.
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330.
- Pezzini, A.; Padovani, A. Lifting the mask on neurological manifestations of COVID-19. Nat. Rev. Neurol. 2020, 16, 636–644.
- Cui, H.; Su, S.; Cao, Y.; Ma, C.; Qiu, W. The Altered Anatomical Distribution of ACE2 in the Brain With Alzheimer’s Disease Pathology. Front. Cell Dev. Biol. 2021, 9, 684874.
- Sardu, C.; Gambardella, J.; Morelli, M.B.; Wang, X.; Marfella, R.; Santulli, G. Hypertension, Thrombosis, Kidney Failure, and Diabetes: Is COVID-19 an Endothelial Disease? A Comprehensive Evaluation of Clinical and Basic Evidence. J. Clin. Med. 2020, 9, 1417.
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860.
- Meinhardt, J.; Radke, J.; Dittmayer, C.; Franz, J.; Thomas, C.; Mothes, R.; Laue, M.; Schneider, J.; Brunink, S.; Greuel, S.; et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat. Neurosci. 2021, 24, 168–175.
- Butowt, R.; Meunier, N.; Bryche, B.; von Bartheld, C.S. The olfactory nerve is not a likely route to brain infection in COVID-19: A critical review of data from humans and animal models. Acta Neuropathol. 2021, 141, 809–822.
- Buzhdygan, T.P.; DeOre, B.J.; Baldwin-Leclair, A.; Bullock, T.A.; McGary, H.M.; Khan, J.A.; Razmpour, R.; Hale, J.F.; Galie, P.A.; Potula, R.; et al. The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood-brain barrier. Neurobiol. Dis. 2020, 146, 105131.
- Khaddaj-Mallat, R.; Aldib, N.; Bernard, M.; Paquette, A.S.; Ferreira, A.; Lecordier, S.; Saghatelyan, A.; Flamand, L.; ElAli, A. SARS-CoV-2 deregulates the vascular and immune functions of brain pericytes via Spike protein. Neurobiol. Dis. 2021, 161, 105561.
- Matschke, J.; Lutgehetmann, M.; Hagel, C.; Sperhake, J.P.; Schroder, A.S.; Edler, C.; Mushumba, H.; Fitzek, A.; Allweiss, L.; Dandri, M.; et al. Neuropathology of patients with COVID-19 in Germany: A post-mortem case series. Lancet Neurol. 2020, 19, 919–929.
- Lee, M.H.; Perl, D.P.; Nair, G.; Li, W.; Maric, D.; Murray, H.; Dodd, S.J.; Koretsky, A.P.; Watts, J.A.; Cheung, V.; et al. Microvascular Injury in the Brains of Patients with COVID-19. N. Engl. J. Med. 2021, 384, 481–483.
- Spudich, S.; Nath, A. Nervous system consequences of COVID-19. Science 2022, 375, 267–269.
- Ye, Q.; Zhou, J.; He, Q.; Li, R.T.; Yang, G.; Zhang, Y.; Wu, S.J.; Chen, Q.; Shi, J.H.; Zhang, R.R.; et al. SARS-CoV-2 infection in the mouse olfactory system. Cell Discov. 2021, 7, 49.
- Zazhytska, M.; Kodra, A.; Hoagland, D.A.; Fullard, J.F.; Shayya, H.; Omer, A.; Firestein, S.; Gong, Q.; Canoll, P.D.; Goldman, J.E.; et al. Disruption of nuclear architecture as a cause of COVID-19 induced anosmia. bioRxiv 2021, 9, 430314.
- Junqueira, C.; Crespo, A.; Ranjbar, S.; de Lacerda, L.B.; Lewandrowski, M.; Ingber, J.; Parry, B.; Ravid, S.; Clark, S.; Schrimpf, M.R.; et al. FcgammaR-mediated SARS-CoV-2 infection of monocytes activates inflammation. Nature 2022, 606, 576–584.
- Chen, Z.; Li, G. Immune response and blood-brain barrier dysfunction during viral neuroinvasion. Innate Immun. 2021, 27, 109–117.
- Jaunmuktane, Z.; Mahadeva, U.; Green, A.; Sekhawat, V.; Barrett, N.A.; Childs, L.; Shankar-Hari, M.; Thom, M.; Jager, H.R.; Brandner, S. Microvascular injury and hypoxic damage: Emerging neuropathological signatures in COVID-19. Acta Neuropathol. 2020, 140, 397–400.
- Achar, A.; Ghosh, C. COVID-19-Associated Neurological Disorders: The Potential Route of CNS Invasion and Blood-Brain Relevance. Cells 2020, 9, 2360.
- Krasemann, S.; Haferkamp, U.; Pfefferle, S.; Woo, M.S.; Heinrich, F.; Schweizer, M.; Appelt-Menzel, A.; Cubukova, A.; Barenberg, J.; Leu, J.; et al. The blood-brain barrier is dysregulated in COVID-19 and serves as a CNS entry route for SARS-CoV-2. Stem. Cell Rep. 2022, 17, 307–320.
- Paniz-Mondolfi, A.; Bryce, C.; Grimes, Z.; Gordon, R.E.; Reidy, J.; Lednicky, J.; Sordillo, E.M.; Fowkes, M. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J. Med. Virol. 2020, 92, 699–702.
- Erickson, M.A.; Rhea, E.M.; Knopp, R.C.; Banks, W.A. Interactions of SARS-CoV-2 with the Blood-Brain Barrier. Int. J. Mol. Sci. 2021, 22, 2681.
- Solomon, I.H.; Normandin, E.; Bhattacharyya, S.; Mukerji, S.S.; Keller, K.; Ali, A.S.; Adams, G.; Hornick, J.L.; Padera, R.F., Jr.; Sabeti, P. Neuropathological Features of COVID-19. N. Engl. J. Med. 2020, 383, 989–992.
- Thakur, K.T.; Miller, E.H.; Glendinning, M.D.; Al-Dalahmah, O.; Banu, M.A.; Boehme, A.K.; Boubour, A.L.; Bruce, S.S.; Chong, A.M.; Claassen, J.; et al. COVID-19 neuropathology at Columbia University Irving Medical Center/New York Presbyterian Hospital. Brain 2021, 144, 2696–2708.
- Frontera, J.A.; Melmed, K.; Fang, T.; Granger, A.; Lin, J.; Yaghi, S.; Zhou, T.; Lewis, A.; Kurz, S.; Kahn, D.E.; et al. Toxic Metabolic Encephalopathy in Hospitalized Patients with COVID-19. Neurocrit. Care 2021, 35, 693–706.
- Taylor, C.T.; Doherty, G.; Fallon, P.G.; Cummins, E.P. Hypoxia-dependent regulation of inflammatory pathways in immune cells. J. Clin. Investig. 2016, 126, 3716–3724.
- Cioffi, M.; Kuffer, J.; Strobel, S.; Dubini, G.; Martin, I.; Wendt, D. Computational evaluation of oxygen and shear stress distributions in 3D perfusion culture systems: Macro-scale and micro-structured models. J. Biomech. 2008, 41, 2918–2925.
- Hayley, S.; Sun, H. Neuroimmune multi-hit perspective of coronaviral infection. J. Neuroinflamm. 2021, 18, 231.
- Amruta, N.; Chastain, W.H.; Paz, M.; Solch, R.J.; Murray-Brown, I.C.; Befeler, J.B.; Gressett, T.E.; Longo, M.T.; Engler-Chiurazzi, E.B.; Bix, G. SARS-CoV-2 mediated neuroinflammation and the impact of COVID-19 in neurological disorders. Cytokine Growth Factor Rev. 2021, 58, 1–15.
- Kanberg, N.; Ashton, N.J.; Andersson, L.M.; Yilmaz, A.; Lindh, M.; Nilsson, S.; Price, R.W.; Blennow, K.; Zetterberg, H.; Gisslen, M. Neurochemical evidence of astrocytic and neuronal injury commonly found in COVID-19. Neurology 2020, 95, e1754–e1759.
- Aschner, M. Immune and inflammatory responses in the CNS: Modulation by astrocytes. Toxicol. Lett. 1998, 102-103, 283–287.
- Graeber, M.B.; Streit, W.J. Microglia: Immune network in the CNS. Brain Pathol. 1990, 1, 2–5.
- Monnet-Tschudi, F.; Zurich, M.G.; Honegger, P. Neurotoxicant-induced inflammatory response in three-dimensional brain cell cultures. Hum. Exp. Toxicol. 2007, 26, 339–346.
- Streit, W.J.; Walter, S.A.; Pennell, N.A. Reactive microgliosis. Prog. Neurobiol. 1999, 57, 563–581.
- Kraft, A.D.; Harry, G.J. Features of microglia and neuroinflammation relevant to environmental exposure and neurotoxicity. Int. J. Environ. Res. Public Health 2011, 8, 2980–3018.
- Claycomb, K.I.; Johnson, K.M.; Winokur, P.N.; Sacino, A.V.; Crocker, S.J. Astrocyte regulation of CNS inflammation and remyelination. Brain Sci. 2013, 3, 1109–1127.
- Dong, Y.; Benveniste, E.N. Immune function of astrocytes. Glia 2001, 36, 180–190.
- Brown, G.C.; Bal-Price, A. Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol. Neurobiol. 2003, 27, 325–355.
- Nakajima, K.; Kohsaka, S. Microglia: Neuroprotective and neurotrophic cells in the central nervous system. Curr Drug Targets Cardiovasc. Haematol. Disord 2004, 4, 65–84.
- Murta, V.; Villarreal, A.; Ramos, A.J. Severe Acute Respiratory Syndrome Coronavirus 2 Impact on the Central Nervous System: Are Astrocytes and Microglia Main Players or Merely Bystanders? ASN Neuro 2020, 12, 1759091420954960.
- Song, E.; Bartley, C.M.; Chow, R.D.; Ngo, T.T.; Jiang, R.; Zamecnik, C.R.; Dandekar, R.; Loudermilk, R.P.; Dai, Y.; Liu, F.; et al. Divergent and self-reactive immune responses in the CNS of COVID-19 patients with neurological symptoms. Cell Rep. Med. 2021, 2, 100288.
- Franke, C.; Ferse, C.; Kreye, J.; Reincke, S.M.; Sanchez-Sendin, E.; Rocco, A.; Steinbrenner, M.; Angermair, S.; Treskatsch, S.; Zickler, D.; et al. High frequency of cerebrospinal fluid autoantibodies in COVID-19 patients with neurological symptoms. Brain Behav. Immun. 2021, 93, 415–419.
- Eden, A.; Kanberg, N.; Gostner, J.; Fuchs, D.; Hagberg, L.; Andersson, L.M.; Lindh, M.; Price, R.W.; Zetterberg, H.; Gisslen, M. CSF Biomarkers in Patients With COVID-19 and Neurologic Symptoms: A Case Series. Neurology 2021, 96, e294–e300.
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405.
- Mohammadi, S.; Moosaie, F.; Aarabi, M.H. Understanding the Immunologic Characteristics of Neurologic Manifestations of SARS-CoV-2 and Potential Immunological Mechanisms. Mol. Neurobiol. 2020, 57, 5263–5275.
- Pereira, A. Long-Term Neurological Threats of COVID-19: A Call to Update the Thinking About the Outcomes of the Coronavirus Pandemic. Front. Neurol. 2020, 11, 308.
- Lu, Y.; Li, X.; Geng, D.; Mei, N.; Wu, P.Y.; Huang, C.C.; Jia, T.; Zhao, Y.; Wang, D.; Xiao, A.; et al. Cerebral Micro-Structural Changes in COVID-19 Patients - An MRI-based 3-month Follow-up Study. EClinicalMedicine 2020, 25, 100484.
- Arachchillage, D.R.J.; Laffan, M. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 1233–1234.
- Lee, S.; Viqar, F.; Zimmerman, M.E.; Narkhede, A.; Tosto, G.; Benzinger, T.L.; Marcus, D.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; et al. White matter hyperintensities are a core feature of Alzheimer’s disease: Evidence from the dominantly inherited Alzheimer network. Ann. Neurol. 2016, 79, 929–939.
- Wen, Y.; Yang, S.; Liu, R.; Brun-Zinkernagel, A.M.; Koulen, P.; Simpkins, J.W. Transient cerebral ischemia induces aberrant neuronal cell cycle re-entry and Alzheimer’s disease-like tauopathy in female rats. J. Biol. Chem. 2004, 279, 22684–22692.
- Gerges Harb, J.; Noureldine, H.A.; Chedid, G.; Eldine, M.N.; Abdallah, D.A.; Chedid, N.F.; Nour-Eldine, W. SARS, MERS and COVID-19: Clinical manifestations and organ-system complications: A mini review. Pathog. Dis. 2020, 78, ftaa033.
- Qi, F.; Qian, S.; Zhang, S.; Zhang, Z. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem. Biophys. Res Commun. 2020, 526, 135–140.
- Sungnak, W.; Huang, N.; Becavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687.
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613.
- Mahalakshmi, A.M.; Ray, B.; Tuladhar, S.; Bhat, A.; Paneyala, S.; Patteswari, D.; Sakharkar, M.K.; Hamdan, H.; Ojcius, D.M.; Bolla, S.R.; et al. Does COVID-19 contribute to development of neurological disease? Immun. Inflamm. Dis. 2021, 9, 48–58.
- Meng, X.; Deng, Y.; Dai, Z.; Meng, Z. COVID-19 and anosmia: A review based on up-to-date knowledge. Am. J. Otolaryngol. 2020, 41, 102581.
- Saniasiaya, J.; Islam, M.A.; Abdullah, B. Prevalence of Olfactory Dysfunction in Coronavirus Disease 2019 (COVID-19): A Meta-analysis of 27,492 Patients. Laryngoscope 2021, 131, 865–878.
- AbdelHamid, S.G.; Refaat, A.A.; Benjamin, A.M.; Elmawardy, L.A.; Elgendy, L.A.; Manolly, M.M.; Elmaksoud, N.A.; Sherif, N.; Hamdy, N.M. Deciphering epigenetic(s) role in modulating susceptibility to and severity of COVID-19 infection and/or outcome: A systematic rapid review. Environ. Sci. Pollut. Res. Int. 2021, 28, 54209–54221.
- Coecke, S.; Munoz, A.; D’Alessandro, V.; De Bernardi, F.; Romeo, P.; Torres, F.; Harris, G.; Parvatam, S. Knowledge from human relevant cell, tissue and mathematics-based methods as key tools for understanding COVID-19. In The Coronavirus Pandemic and the Future: Virology, Epidemiology, Translational Toxicology and Therapeutics; Waters, M.D., Dhawan, A., Marrs, T., Anderson, D., Warren, S., Hughes, C.L., Eds.; Royal Society of Chemistry: London, UK, 2021; pp. 452–455. Available online: https://www.chemistryworld.com/the-coronavirus-pandemic-and-the-future/knowledge-from-human-relevant-cell-tissue-and-mathematics-based-methods-as-key-tools-for-understanding-covid-19/4013732.article (accessed on 13 September 2022).
- Bilinska, K.; Jakubowska, P.; Von Bartheld, C.S.; Butowt, R. Expression of the SARS-CoV-2 Entry Proteins, ACE2 and TMPRSS2, in Cells of the Olfactory Epithelium: Identification of Cell Types and Trends with Age. ACS Chem. Neurosci. 2020, 11, 1555–1562.
- Zazhytska, M.; Kodra, A.; Hoagland, D.A.; Frere, J.; Fullard, J.F.; Shayya, H.; McArthur, N.G.; Moeller, R.; Uhl, S.; Omer, A.D.; et al. Non-cell-autonomous disruption of nuclear architecture as a potential cause of COVID-19-induced anosmia. Cell 2022, 185, 1052–1064.e12.
- Prem, B.; Liu, D.T.; Besser, G.; Sharma, G.; Dultinger, L.E.; Hofer, S.V.; Matiasczyk, M.M.; Renner, B.; Mueller, C.A. Long-lasting olfactory dysfunction in COVID-19 patients. Eur. Arch. Otorhinolaryngol. 2022, 279, 3485–3492.
- SARS-CoV-2 Infection of Olfactory Epithelium Leading to Impaired Olfactory Function (Short-Term Anosmia). Available online: https://aopwiki.org/aops/394 (accessed on 13 September 2022).
- Shahbaz, M.A.; De Bernardi, F.; Alatalo, A.; Sachana, M.; Clerbaux, L.-A.; Muñoz, A.; Parvatam, S.; Landesmann, B.; Kanninen, K.M.; Coecke, S. Mechanistic Understanding of the Olfactory Neuroepithelium Involvement Leading to Short-Term Anosmia in COVID-19 Using the Adverse Outcome Pathway Framework. Cells 2022, 11, 3027.
- Wu, Y.; Xu, X.; Chen, Z.; Duan, J.; Hashimoto, K.; Yang, L.; Liu, C.; Yang, C. Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav. Immun. 2020, 87, 18–22.
- Iaconetta, G.; De Luca, P.; Scarpa, A.; Cassandro, C.; Cassandro, E. Meningoencephalitis Associated with SARS-Coronavirus-2. Transl. Med. UniSa 2020, 23, 42–47.
- Moriguchi, T.; Harii, N.; Goto, J.; Harada, D.; Sugawara, H.; Takamino, J.; Ueno, M.; Sakata, H.; Kondo, K.; Myose, N.; et al. A first case of meningitis/encephalitis associated with SARS-Coronavirus-2. Int. J. Infect. Dis. 2020, 94, 55–58.
- Xiang, Y.T.; Zhao, Y.J.; Liu, Z.H.; Li, X.H.; Zhao, N.; Cheung, T.; Ng, C.H. The COVID-19 outbreak and psychiatric hospitals in China: Managing challenges through mental health service reform. Int. J. Biol. Sci. 2020, 16, 1741–1744.
- Vargas, G.; Medeiros Geraldo, L.H.; Gedeao Salomao, N.; Viana Paes, M.; Regina Souza Lima, F.; Carvalho Alcantara Gomes, F. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and glial cells: Insights and perspectives. Brain Behav. Immun. Health 2020, 7, 100127.
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145.
- Chatterjee, D.; Biswas, K.; Nag, S.; Ramachandra, S.G.; Das Sarma, J. Microglia play a major role in direct viral-induced demyelination. Clin. Dev. Immunol. 2013, 2013, 510396.
- Wheeler, D.L.; Sariol, A.; Meyerholz, D.K.; Perlman, S. Microglia are required for protection against lethal coronavirus encephalitis in mice. J. Clin. Investig. 2018, 128, 931–943.
- Binding of Sars-CoV-2 Spike Protein to ACE 2 Receptors Expressed on Brain Cells (Neuronal and Non-Neuronal) Leads to Neuroinflammation Resulting in Encephalitis. Available online: https://aopwiki.org/aops/374 (accessed on 13 September 2022).
- Montalvan, V.; Lee, J.; Bueso, T.; De Toledo, J.; Rivas, K. Neurological manifestations of COVID-19 and other coronavirus infections: A systematic review. Clin. Neurol. Neurosurg. 2020, 194, 105921.
- Aghayari Sheikh Neshin, S.; Shahjouei, S.; Koza, E.; Friedenberg, I.; Khodadadi, F.; Sabra, M.; Kobeissy, F.; Ansari, S.; Tsivgoulis, G.; Li, J.; et al. Stroke in SARS-CoV-2 Infection: A Pictorial Overview of the Pathoetiology. Front. Cardiovasc. Med. 2021, 8, 649922.
- Divani, A.A.; Andalib, S.; Di Napoli, M.; Lattanzi, S.; Hussain, M.S.; Biller, J.; McCullough, L.D.; Azarpazhooh, M.R.; Seletska, A.; Mayer, S.A.; et al. Coronavirus Disease 2019 and Stroke: Clinical Manifestations and Pathophysiological Insights. J. Stroke Cerebrovasc. Dis. 2020, 29, 104941.
- Zhou, Y.L.; Lu, J.; Cheng, Y.B.; Xin, N. Nervous system complications of COVID-19 with a focus on stroke. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 13044–13048.
- Binding of Sars-CoV-2 Spike Protein to ACE 2 Receptors Expressed on Pericytes Leads to Disseminated Intravascular Coagulation Resulting in Cerebrovascular Disease (Stroke). Available online: https://aopwiki.org/aops/395 (accessed on 13 September 2022).
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J., Jr.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE official report: A practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482.
- Vitalakumar, D.; Sharma, A.; Kumar, A.; Flora, S.J.S. Neurological Manifestations in COVID-19 Patients: A Meta-Analysis. ACS Chem. Neurosci. 2021, 12, 2776–2797.
- Nikbakht, F.; Mohammadkhanizadeh, A.; Mohammadi, E. How does the COVID-19 cause seizure and epilepsy in patients? The potential mechanisms. Mult. Scler. Relat. Disord. 2020, 46, 102535.
- Kwon, A.; Kwak, B.O.; Kim, K.; Ha, J.; Kim, S.J.; Bae, S.H.; Son, J.S.; Kim, S.N.; Lee, R. Cytokine levels in febrile seizure patients: A systematic review and meta-analysis. Seizure 2018, 59, 5–10.
- Swissa, E.; Serlin, Y.; Vazana, U.; Prager, O.; Friedman, A. Blood-brain barrier dysfunction in status epileptics: Mechanisms and role in epileptogenesis. Epilepsy Behav. 2019, 101, 106285.
- de Vries, E.E.; van den Munckhof, B.; Braun, K.P.; van Royen-Kerkhof, A.; de Jager, W.; Jansen, F.E. Inflammatory mediators in human epilepsy: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2016, 63, 177–190.
- Kovacs, R.; Heinemann, U.; Steinhauser, C. Mechanisms underlying blood-brain barrier dysfunction in brain pathology and epileptogenesis: Role of astroglia. Epilepsia 2012, 53 (Suppl. S6), 53–59.
- Asadi-Pooya, A.A.; Simani, L.; Shahisavandi, M.; Barzegar, Z. COVID-19, de novo seizures, and epilepsy: A systematic review. Neurol. Sci. 2021, 42, 415–431.
- Lau, K.K.; Yu, W.C.; Chu, C.M.; Lau, S.T.; Sheng, B.; Yuen, K.Y. Possible central nervous system infection by SARS coronavirus. Emerg. Infect. Dis. 2004, 10, 342–344.
- Hung, E.C.; Chim, S.S.; Chan, P.K.; Tong, Y.K.; Ng, E.K.; Chiu, R.W.; Leung, C.B.; Sung, J.J.; Tam, J.S.; Lo, Y.M. Detection of SARS coronavirus RNA in the cerebrospinal fluid of a patient with severe acute respiratory syndrome. Clin. Chem. 2003, 49, 2108–2109.
- Villeneuve, D.L.; Crump, D.; Garcia-Reyero, N.; Hecker, M.; Hutchinson, T.H.; LaLone, C.A.; Landesmann, B.; Lettieri, T.; Munn, S.; Nepelska, M.; et al. Adverse outcome pathway (AOP) development I: Strategies and principles. Toxicol. Sci. 2014, 142, 312–320.
- Villeneuve, D.L.; Angrish, M.M.; Fortin, M.C.; Katsiadaki, I.; Leonard, M.; Margiotta-Casaluci, L.; Munn, S.; O’Brien, J.M.; Pollesch, N.L.; Smith, L.C.; et al. Adverse outcome pathway networks II: Network analytics. Environ. Toxicol. Chem. 2018, 37, 1734–1748.
This entry is offline, you can click here to edit this entry!