Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
|
Biochemistry & Molecular Biology
The tumor suppressor p53, encoded by the TP53 gene and known as “the guardian of the genome”, performs a variety of functions in cancer prevention.
- mutant p53
- gain-of-function
- targeted therapy
- p53 reactivation
1. Introduction
The tumor suppressor p53, encoded by the TP53 gene and known as “the guardian of the genome” [1], performs a variety of functions in cancer prevention. The basic unit of the p53 protein consists of three major functional domains such as an N-terminal transactivation domain (TAD), a core DNA-binding domain (DBD)—the main target for mutations, and a C-terminal regulatory domain (CTD) (Figure 1A) [2]. The p53 protein, to exert its function, binds in a sequence-specific manner to the DNA-binding sites by forming a tetramer, via four self-assembling p53 molecules, which are stabilized by protein–protein and base-stacking interactions [3]. As a gene-expression regulator, it mainly controls how the cell behaves under stress conditions. p53-specific responses consist of the activation of mechanisms such as cell cycle arrest, apoptosis, and senescence [4,5,6,7]. Responsible for such pivotal processes, p53 can deliver considerable damage if mutated, thus becoming “the guardian of the cancer cell” [8]. TP53 is the most frequently mutated gene in cancers. It has been recognized that 50% of cancer patients acquire certain types of TP53 gene alterations [9,10]. The potential role of p53 as a specific target in modern therapies against cancers is being widely discussed. Numerous attempts at this approach have already been made. TP53 mutations are labeled as loss of function (LOF) or gain of function (GOF). The presence of GOF TP53 mutants increases the malignancy of tumors in various ways. Occurring metastases, greater chemoresistance, invasiveness, and shorter survival are typical traits of GOF. These mutations account for 30% of all missense mutations in the TP53 gene. Widespread screening of patients allowed for the recognition of the hotspots R175, G245, R248, R249, R273, and R282, which are present in the DNA-binding domain (DBD) or near this interface of p53 [11].
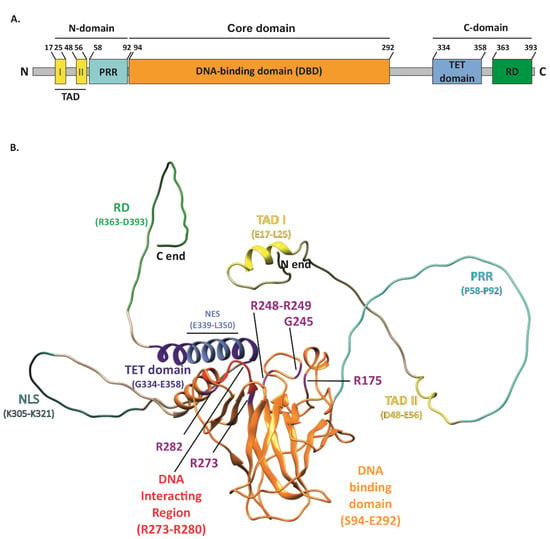
Figure 1. Structure of the p53 protein. (A) Simplified representation of the secondary structure showing domain organization of the human p53 protein (Uniprot #P04637). TAD, transactivation domain; PRR, proline-rich region; DBD, DNA-binding domain; TET, tetramerization domain; RD, regulatory domain. (B) Schematic representation of the p53 3D structure with the GOF mutation sites shown (purple). The TAD domain is shown in yellow with TAD I and II motifs indicated (yellow); PRR (cyan); DBD (orange) with direct DNA-binding region indicated (red); bipartite nuclear localization signal (NLS) (sea green); TET domain (dark blue) including nuclear export signal (NES) (light blue); and C-terminal RD (green). The structure of p53 was only partially solved by crystallography/X-ray diffraction or NMR; therefore, the 3D structure prediction of full-length protein was performed with AlphaFold DB, DeepMind Technologies Limited [12,13], and visualized with UCSF Chimera, an extensible molecular modeling system developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco [14].
2. Pivotal Functions and Regulation Mechanisms of Wild-Type p53 (WTp53)
Non-altered p53 acts as a transcription regulator in charge of the cellular responses to stress factors, hypoxia, nutritional stress, differentiation signals, and DNA damage. Under stress conditions, an affected cell can respond in numerous ways. p53 induces cell cycle arrest, apoptosis, or senescence. Each of those processes is regulated by p53 and its target genes. Cell cycle arrest is induced by p21 and p57; apoptosis is activated via Puma, also known as BCL2 binding component 3, Bax (BCL2-associated X), and Noxa (phorbol-12-myristate-13-acetate-induced protein 1); and senescence is induced via p27Kip1 (cyclin-dependent kinase inhibitor 1B, encoded by CDKN1B gene) and Pai1 (phosphoribosylanthranilate isomerase 1), while TIGAR (TP53 induced glycolysis regulatory phosphatase) and glutaminase 2 (encoded by GLS2) are responsible for the arrangement of metabolic changes [7,15,16]. Additionally, p53 upregulates the expression of death receptor DR5 and, thus, may mediate apoptosis in part via DR5 [17]. The negative regulation of p53 activity is dependent on the Mdm2 protein, which binds to p53 and, as a result, inhibits its transcriptional functions, thus promoting proteasomal degradation. WTp53 oncogenic suppressor functions are regulated by the presence of molecular chaperones, which shape the proper tertiary and quaternary structure of the protein [18,19].
However, mutant p53 (MUTp53) benefits from this mechanism, due to the chaperones that stabilize MUTp53, thus allowing it to escape proteasomal degradation [20]. It is a well-known fact that MUTp53 is abundant in cancer cells compared to WTp53 in non-tumorigenic cells, which indicates that the mutated protein is more stable [21]. Although MUTp53 undergoes the degradation mechanisms orchestrated by Mdm2 in normal tissues, the same process fails in tumors for an unknown reason [22]. Several studies were conducted to explain this phenomenon. Cancer tissues have the tendency to express HSP70 or HSP90, which stabilize MUTp53, therefore allowing its accumulation in the cell [23]. However, this explanation undermines the regulatory patterns, including those controlled by Mdm2 or other E3 ligase proteins [24].
3. Features of GOF p53 Mutants
Missense mutations occurring in the six hot spots consist of eight mutants, which account for nearly 30% of all missense mutations. They consist of R175H, G245S, R248Q, R248W, R249S, R273C, R273H, and R282W. The gain of function characteristics are designated to all of them, although the mechanisms behind their novel functionalities are different. This group can be further divided, based on the dysfunctionality that is present in the final protein. Mutation in the contact mutants, R248Q, R248W, R273H, and R273C, occurs in their DNA-binding domain, directly affecting their ability to control the transcription of targeted genes. Conformational mutants such as R175H, G245S, R249S, and R282H, on the other hand, are unable to fold properly, leading to the loss of the zinc coordinates and, thus, general DNA-binding activity [25,26,27,28,29,30]. Contact mutants and conformational (structure) mutants show decreased thermostability. Altered proteins are not capable of binding to the designated sites; however, they are capable of binding to new sequences, can regulate completely different genes, as a result, and produce new phenotypes [30]. Consequently, each of the substitutions can affect the newly formed protein differently; hence, each mutant promotes distinct GOF hallmarks caused by individual molecular mechanisms, which may require a novel approach in therapy. The clinical approach is highly complicated, because many factors must be taken into account; nevertheless, the result could notably benefit the condition of cancer patients [7,31,32]. More than 40% of cancer-related gene expression depends on the SWI/SNF signaling pathway. SWI/SNF is a subfamily of ATP-dependent chromatin remodeling complexes, which serve broad roles in the transcriptional regulation of differentiation and proliferation across many lineages. Lately, it has been shown that MUTp53 can interact with the SWI/SNF complex, resulting in chromatin being in an open state due to the histone modifications. As a consequence, changes in the expression of cancer-related genes were observed [33]. Other proteins undergo changes in their activity via MUTp53, such as p63 and p73 [34], which, in turn, inhibit apoptosis instead of inducing it. Various other transcription factors are under the influence of MUTp53 both in negative and positive regulatory activity: ETS2, NF-kB (nuclear factor kappa B), HIF-1α (hypoxia-inducible factor 1 subunit alpha), SMAD, SREBP (sterol regulatory element binding protein), and NF-Y (nuclear transcription factor Y) [29].
4. Chemoresistance Mechanisms Established by MUTp53
Cancer cells’ ability to manage oxidative stress is possible due to the presence of the xCT (also known as SLC7A11), a functional subunit of the cystine/glutamate antiporter system xc-. xCT is coded by the SLC7A11 (solute carrier family 7, member 11) gene, and its overexpression is observed in various types of cancer cells, especially in cancer stem cells (CSC). xCT alters metabolic pathways via participation in glutathione biosynthesis and, in this way, protects cancer cells from oxidative stress conditions and ferroptosis [35]. Generally, the presence of xCT in cells is associated with the promotion of tumor progression and induction of chemoresistance through the detoxification activity of GSH-mediated reactive oxygen species (ROS). It was shown that WTp53- as well as p53-carrying GOF missense mutations can inhibit the expression of SLC7A11 and sensitize cells to ferroptosis. It was also shown that cells with non-functional p53 are highly resistant to chemotherapeutics and radiotherapy; however, the knockout of the SLC7A11 gene results in the restoration of sensitivity to applied therapy. These observations are related to GSH depletion and, consequently, to the reduced protection from oxidative stress upon xCT inhibition. This suggests that, in the resistance to oxidative stress, the regulation of the xCT-glutathione axis plays an important role, which allows the tumor to survive in unsuitable conditions. Therefore, the inhibition of the xCT-glutathione axis may represent a promising approach to overcoming resistance associated with MUTp53 [36,37]. This significantly increases the resistance to therapeutic drugs, such as cisplatin, adriamycin, and etoposide, even when compared to cancer cell lines with TP53 knockdown [38]. Etoposide resistance was also observed in the study conducted by Scian et al., where it was linked to the increased expression of NF-κB2 induced by MUTp53R273H and R175H. Interestingly, the mutant D281G did not cause such effects [39]. Cisplatin resistance can be overcome in the mutant R273H via depletion of ataxia-telangiectasia (ATM) and the Rad3-related protein (ATR) activator DNA2 [40]. The understanding of these mechanisms is crucial for the successful treatment of patients. Overall, studies show that MUTp53 is involved in the increased expression of MDR1 (multidrug resistance gene 1) [41]. Each type of GOF MUTp53 can manifest several chemoresistance mechanisms due to different proteins and genes being affected. For instance, R273H is resistant to doxorubicin and methotrexate via the inhibition of apoptosis through procaspase-3 downregulation [42]. The resistance to gemcitabine occurs due to MUTp53 phosphorylation, which induces CDK1 (cyclin-dependent kinase 1) and CCNB1 (cyclin B1) expression [43], while R273H mutant is resistant to cisplatin via YAP/β-arrestin1 pathway [44]. Long non-coding RNAs (lncRNAs) are associated with chemoresistance and the proliferation of tumors. Cells carrying the R273H mutation were established to have more in common with CSC than other mutants. Moreover, lnc273-31 and lnc273-34 were required for CSC to establish the self-renewal feature. Generally, epithelial–mesenchymal transition (EMT), migration, invasion, and chemoresistance were established as the characteristics of R273H mutant cells, which demonstrate the high expression of lnc273-31 and lnc273-34. However, this effect was not manifested in R175H or R248W p53 mutants [45]. Another noteworthy chemoresistance mechanism involves the Mdm2-mediated ubiquitination and degradation of the mutant p53. It was observed that the p53R248Q mutant’s resistance to cisplatin could be modulated by fibroblast growth factor-inducible 14 (Fn14). High-grade serous ovarian cancer cells became sensitive to cisplatin, due to p53R248Q degradation, which was possible when expression of Fn14 was restored [46].
This entry is adapted from the peer-reviewed paper 10.3390/ijms232113287
This entry is offline, you can click here to edit this entry!