Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Terpenoids, also known as isoprenoids, are one of the largest natural product families, constituting more than 40,000 primary and secondary metabolites, including monoterpenes (53%), diterpenoids (1%), sesquiterpenes (28%), and others (18%). The basic unit of terpenes is the isoprene unit (C5H8), which is a simple hydrocarbon. It is the main precursor and could be post-modified through the cytosolic mevalonate (MVA) pathway or the plastid methyl erythritol phosphate (MEP) pathway. Terpenoids are a major source of bioactive natural products.
- phytochemicals
- antimicrobial agents
- biosynthetic pathway
1. The Antimicrobial Mechanisms of Terpenoids
There are mainly five mechanisms through which terpenoids exhibit antimicrobial action according to previous reports.
-
Cell membrane destruction: Terpenoids mainly use their lipophilicity to destroy the cell membrane of bacteria. Terpenoids can pass through the phospholipid bilayer of bacteria and diffuse inward, showing antibacterial or bactericidal effects [1]. Since the integrity of the cell membrane is very important for the normal physiological activities of bacteria, the damage of terpenoids to the membrane will affect the bacteria’s basic physiological activities, and the important substances such as proteins and important enzymes in the cell will be lost, finally achieving the antimicrobial effect [2]. It is reported that 1,8-cineole (Table 1), a monoterpene substance extracted from Eucalyptus globulus Labill, showed antibacterial effect against Acinetobacter baumannii, Candida albicans, a methicillin-resistant Staphylococcus aureus (MRSA) strain, and Escherichia coli by destroying the cell membrane [3]. In another study, the researchers exposed Salmonella typhimurium, E. coli O157: H7, Pseudomonas fluorescence, Brochotrix thermophacta, and Staphylococcus aureus cells to cinnamaldehyde (Table 1), carvacrol (Table 1), thymol (Table 1), eugenol (Table 1), and limonene (Table 1), and observed their membrane damage through scanning electron microscopy. These results found that terpenoids can achieve bacteriostatic effects by destroying the membrane structure [4]. The mechanism of action and target sites on microbial cells are graphically illustrated in [1][2].
-
Anti-quorum sensing (QS) action: The QS system is an intercellular communication system [1]. It is a communication mode for bacteria to coordinate the interaction between bacteria and other organisms, which is also the main reason for the emergence of antibiotic resistance [5]. The group sensing signal loop of Gram-positive and Gram-negative bacteria has been introduced and illustrated in the literature [6]. Studies have shown that a low concentration of cinnamaldehyde can effectively inhibit the QS effect between bacteria [7]. Low concentrations of carvacrol and thymol can effectively inhibit the self-inducer of bacteria, namely, acyl homoserine lactone (AHL), thus achieving the inhibition of QS [8]. The action mechanism of cinnamaldehyde inhibiting the acyl homoserine lactones and other autoinducers involved in the quorum sensing is illustrated in [9].
-
Inhibition of ATP and its enzyme: ATP is the most direct energy source in organisms, and it is also a necessary element for microorganisms to maintain normal operation and work. Terpenoids can act on the cell membrane, resulting in the difference in ATP concentration inside and outside the cell, leading to the disorder of the cell membrane, thus conducting the antibacterial activity [1]. For example, terpenoid eugenol and thymol could target the cell membrane to show fungicidal activity against C. albicans by inhibiting H+-ATPase, which will lead to intracellular acidification and cell death [10]. In another study, the researchers treated the target pathogen with the MIC of carvacrol. The extracellular ATP concentrations of the samples were measured with the help of a luminometer (Biotek). On the basis of absorbance analysis at 260 nm, this study observed that carvacrol disrupted the E. coli membrane, while the release of potassium ions and ATP was also detected [11].
-
Inhibition of protein synthesis: The physiological activity of bacteria is inseparable from protein synthesis. Terpenoids, as inhibitors of protein synthesis, can achieve an antibacterial effect by blocking any process of the protein synthesis pathway. Some studies have shown that cinnamaldehyde can reduce the in vitro assembly reaction and the binding reaction of FtsZ (filamenting temperature-sensitive mutant Z)-type protein, a prokaryotic homolog of tubulin that regulates cell division. In addition, cinnamaldehyde can inhibit the hydrolysis of GTP and bind to FtsZ, as well as interfere with the formation of z-loop of cell dynamics, thus showing antibacterial activity against bacteria [12]. In the latest research, the researchers used calculations, biochemistry, and in-vivo-based assays to verify that cinnamaldehyde is a potential inhibitor of S. typhimurium (stFtsZ), and its inhibition rate of stFtsZ GTPase activity and polymerization is up to 70% [13].
-
The synergistic effect: For example, the synergistic antibacterial effect of eugenol with carvacrol and thymol is due to the ability of carvacrol and thymol to penetrate the extracellular membrane, thus making it easier for eugenol to enter the cytoplasmic membrane or increasing the number, size, and duration of pores to bind to membrane proteins for better antibacterial activity [14]. The reaction mechanism is shown in the literature [9].
Table 1. Summary of the antimicrobial effects of some plant-derived terpenoids, alkaloids, and flavonoids.
| Compounds | Chemical Structures | Target Microorganisms | Antimicrobial Effects | Reference | |
|---|---|---|---|---|---|
| Terpenoids | 1,8-cineole | 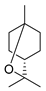 |
A. baumannii C. albicans MRSA strain E. coli |
Cell membrane destruction | [3] |
| cinnamaldehyde | 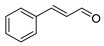 |
S. typhimurium E. coli O157: H7 P. fluorescence B. thermophacta S. aureus |
1. Cell membrane destruction 2. Anti-quorum sensing action 3. Inhibition of protein synthesis |
[4][7][12][13] | |
| carvacrol | 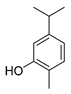 |
S. typhimurium E. coli O157: H7 P. fluorescence B. thermophacta S. aureus P. fluorescens KM121 |
1. Cell membrane destruction 2. Anti-quorum sensing action 3. Inhibition of nucleic acid synthesis 4. The synergistic effect 5. Inhibits cell movement and bacterial invasion |
[4][8][9][11][14] | |
| thymol | 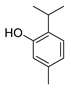 |
S. typhimurium E. coli O157: H7 P. fluorescence B. thermophacta S. aureus P. fluorescens KM121 |
1. Cell membrane destruction 2. Anti-quorum sensing action 3. Inhibition of nucleic acid synthesis 4. The synergistic effect |
[4][8][9][10][14] | |
| eugenol | 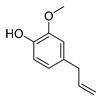 |
S. typhimurium E. coli O157: H7 P. fluorescence B. thermophacta S. aureus |
1. Cell membrane destruction 2. Inhibition of nucleic acid synthesis 3. The synergistic effect |
[4][9][10][14] | |
| limonene | 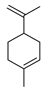 |
A. baumannii C. albicans MRSA strain E. coli |
Cell membrane destruction | [4] | |
| oleanolic acid | 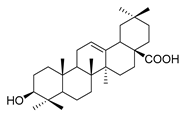 |
E. coli S. aureus Enterococcus faecalis P. aeruginosa |
Antibacterial | [15] | |
| Alkaloids | piperine | 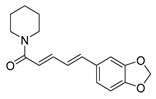 |
S. aureus B. subtilis Salmonella sp. E. coli |
Efflux pump inhibition | [16][17] |
| reserpine | 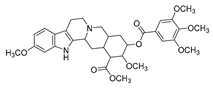 |
E. coli | Efflux pump inhibition | [18] | |
| berberine | 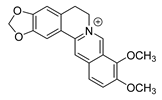 |
E. coli Micrococcus luteus P. aeruginosa Prevotella intermedia Fusobacterium nucleatum MRSA strain |
1. Efflux pump inhibition 2. DNA-intercalating 3. Growth inhibition |
[19][20][21] | |
| L-ephedrine | 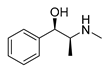 |
Influenza A virus | DNA-intercalating | [22] | |
| D-pseudoephedrine | 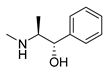 |
Influenza A virus | DNA-intercalating | [22] | |
| L-methylephedrine | 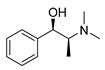 |
Influenza A virus | DNA-intercalating | [22] | |
| chelerythrine | 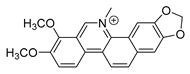 |
S. aureus MRSA strain ESBLs-SA |
1. Nucleic acid synthesis and repair inhibition 2. Growth inhibition |
[23] | |
| 8-hydroxy quinoline | 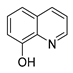 |
S. aureus H. influenza S. pneumoniae |
Permeability change of membrane | [24][25] | |
| michellamine b | 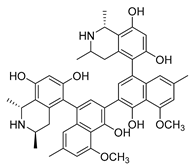 |
HIV | Protein activity inhibition | [26] | |
| sanguinarine | 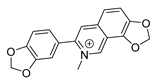 |
K. pneumoniae MRSA strain P. aeruginosa Streptococcus pyogenes |
1. DNA-intercalating 2. Growth inhibition |
[27][28] | |
| roemerine | 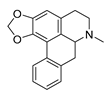 |
S. aureus B. subtilis |
1. Efflux pump inhibition 2. Permeability change of membrane |
[29][30] | |
| dihydrochelerythrine | 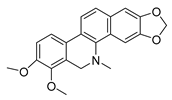 |
S. aureus MRSA strain |
Growth inhibition | [31] | |
| evodiamine | 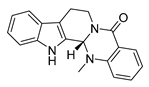 |
M. tubercolosis | Peptidoglycan biosynthesis inhibitor | [32][33] | |
| Flavonoids | hesperidin | 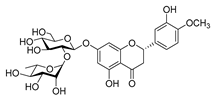 |
S. aureus L. monocytogenes |
Inhibit bacterial growth by modulating the expression of virulence factors | [34] [35] |
| oroxylin a | 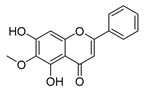 |
B. subtilis S. aureus |
/ | [36] | |
| apigenin | 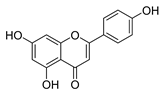 |
S. aureus B. subtilis E. coli P. aeruginosa. |
1. Inhibits peptidoglycan synthesis 2. Increases cell membrane permeability |
[37] | |
| morin | 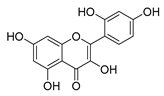 |
E. coli | Inhibition of ATP synthetase | [38] | |
| silymarin | 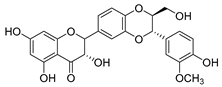 |
E. coli | Inhibition of ATP synthetase | [38] | |
| epigallocatechin gallate | 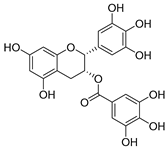 |
S. maltophilia | Inhibits dihydrofolate reductase | [39] | |
| quercetin | 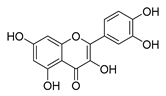 |
P. aeruginosa | 1. Inhibits viral polymerase and viral nucleic acid 2. Inhibits the formation of its biofilm |
[40] | |
| galangin | 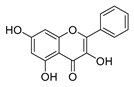 |
S. aureus | 1. Destroys the plasma membrane 2. Weakens the cell wall |
[41] | |
| catechin | 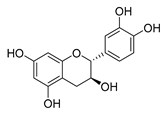 |
B. subtilis E. coli |
Inhibits the bacterial DNA gyrase | [42] [43] |
|
| baicalin | 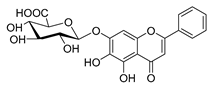 |
Salmonella spp. Staphylococcus spp. |
Inhibits biofilm formation | [44] [45] |
|
| phloretin | 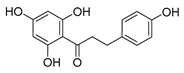 |
C. albicans | 1. Inhibits the pathogenicity 2. Inhibits virulence factors |
[46] | |
| silybin | 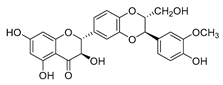 |
MRSA strain | Inhibits the efflux pump | [47] |
2. Biosynthesis of Terpenoid Precursors
There are two important precursors for terpenoid biosynthesis, dimethylallyl pyrophosphates (DMAPP) and isopentenyl diphosphate (IPP), which can both be produced via the MVA or MEP pathways (Figure 1), depending on the organism. The MVA pathway is based on the formation of IPP and DMAPP from acetyl coenzyme A (CoA) through the precursor substance MVA, followed by further condensation of IPP and DMAPP to form sesquiterpenes, triterpenes, and sterols by the action of polyisoprene pyrophosphate synthase. The MEP pathway, on the other hand, is based on pyruvate and glyceraldehyde-3-phosphate in the presence of 1-deoxyxylulose-5-phosphate synthase (DXS) to form DXP. Then, under the catalysis of 1-deoxyxylulose-5-phosphate reductor isomerase (DXR), MEP was formed, followed by further phosphorylation and cyclization to produce IPP, which will be used in the downstream biosynthesis of monoterpenes, diterpenes, and other terpenoids. In plants, both pathways can occur, with the MVA pathway acting in the cytoplasm and the MEP pathway acting in the plastid. In bacteria, terpenoids are generally produced via the MEP pathway, whereas terpenoids are mostly synthesized via the MVA pathway in fungi. Although there are slight differences in the processes of these two pathways, the end products are both DMAPP and IPP [48]. In general, the MEP pathway provides C5-pentenyl diphosphate for the synthesis of C10 monoterpenes, C20 diterpenes, and C40 tetraterpenes, while the MVA pathway provides the same generic precursors for the synthesis of C15 sesquiterpenes, C27–29 sterols, C30 triterpenes, and their saponin derivatives [49].
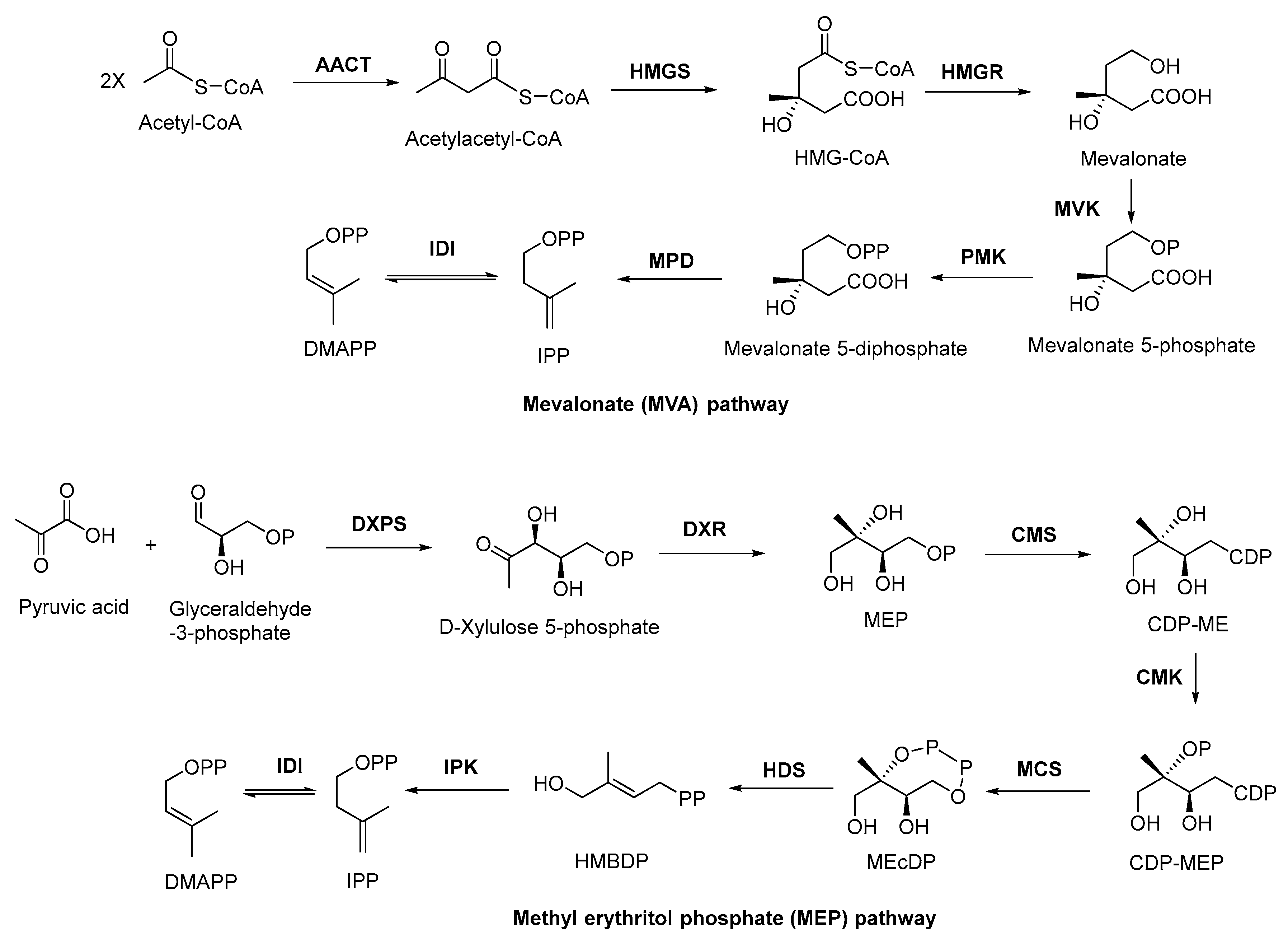
Figure 1. MVA and MEP pathways involved in terpenoid biosynthesis. AACT, acetoacetyl coenzyme A thiolase; HMGS, 3-hydroxy-3-methyl glutaryl coenzyme A synthetase; HMGR, 3-hydroxy-3-methyl glutaryl coenzyme A reductase; MVK, mevalonate kinase; PMK, phosphomevalonate kinase; MPD, mevalonate-5-pyrophosphate decarboxylase; IDI, isopentenyl diphosphate isomerase; DXPS, 1-deoxy-xylose-5-phosphate synthase; DXR, 1-deoxy-xylose-5-phosphate racemic enzyme; CMS, 4-diphoxphocyt-idyl-2-C-methyl-2-(E)-butenyl-4-diphosphate synthase; CDP, cytidine-4-diphosphate; CDP-ME, cytidine-4-diphosphate-2-C-methylerythritol; CMK, 4-diphoxphocyt-idyl-2-C-methyl-D-erythritol kinase; CDP-MEP, cytidine-4-diphosphate-2-C-methyl-D-erythritol-2-phosphate; MCS, 2-C-methyl-D-erythritol-2,4-cyclodiphosphats synthase; MEcDP, 2-C-methyl-D-erythritol-2,4-cyclophosphoric acid; HDS, 1-hydroxy-2-methyl-2-(E)-butenyl-4-diphosphate synthase; HMBDP, 1-hydroxy-2-methyl-2-(E)-butenyl-4-diphosphate; IPK, isopentenyl monophosphate kinase.
3. Discovery, Biosynthesis Investigations, and Engineering Strain Construction of the Representative Terpenoid Antimicrobial Agent—Artemisinin
3.1. Discovery and Predicted Action Mechanism of Artemisinin
So far, there have been several reports about terpenoid compounds that displayed desired antimicrobial activities [50]. Among them, the most representative one is undoubtedly artemisinin (Figure 2). Artemisinin (Qinghaosu) is a sesquiterpene endoperoxide isolated from the leaves of the plant Artemisia annua, which has a long history of use in traditional Chinese medicine. Malaria, caused by Plasmodium falciparum, has been a life-threatening disease for thousands of years [51]. Nowadays, 40% of the world’s population is at risk of malaria infection, and artemisinin is designated as the first-line antimalarial drug by the World Health Organization (WHO). Since the discovery of the antimalarial activities of artemisinin by Chinese scientists in 1971, it has saved millions of lives and represents one of the significant contributions of China to global health. On account of this, the 2015 Nobel Prize for Medicine was awarded to Professor Youyou Tu for her contributions to the discovery and recognition of artemisinin [52].
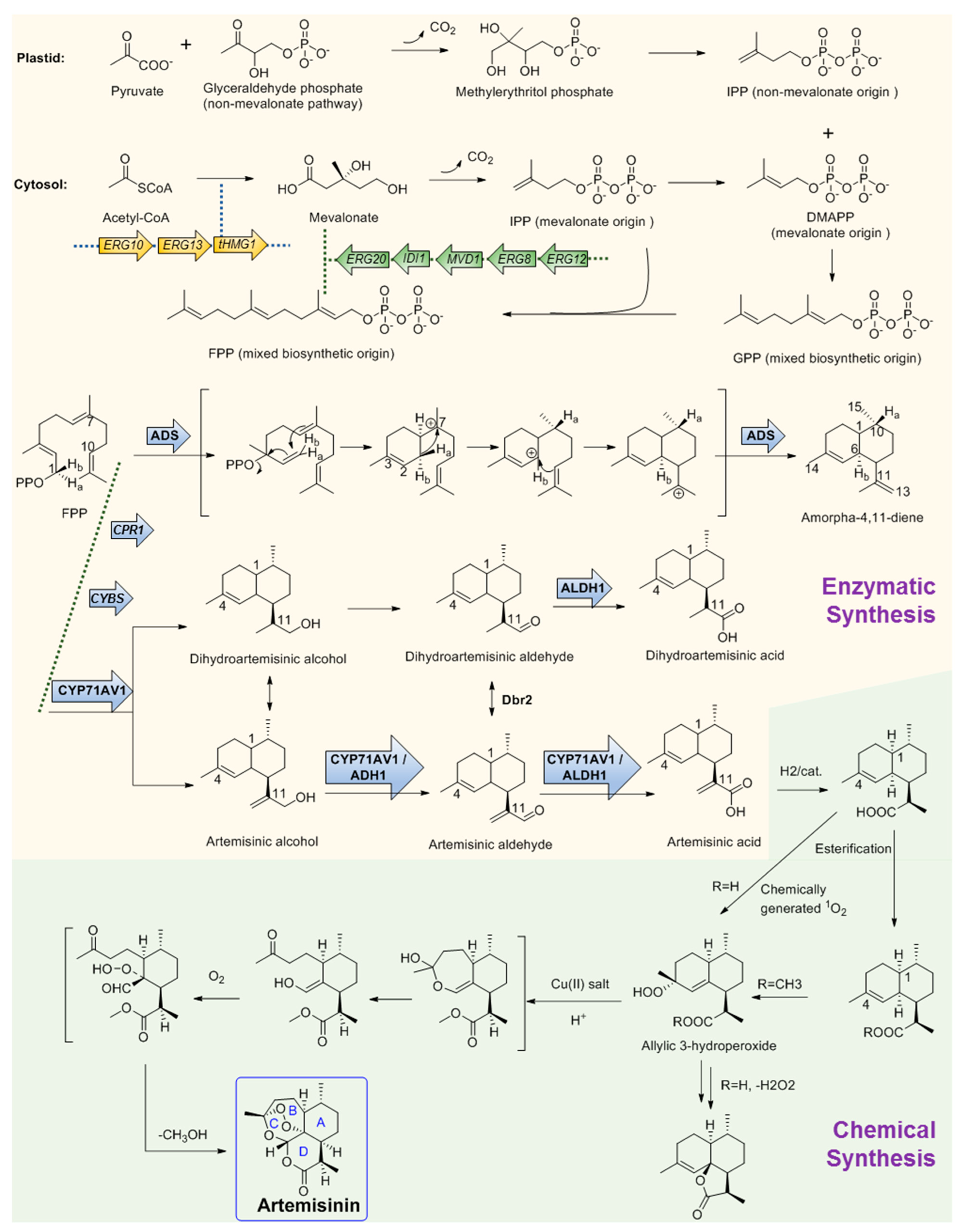
Figure 2. Chemo-enzymatic synthesis of artemisinin. Yellow region shows the biosynthesis pathways for artemisinic acid production. Green region shows the chemical conversion route of artemisinic acid to artemisinin. tHMGR, 3-hydroxy-3-methylglutaryl-coenzyme A reductase; ADS, amorphadiene synthase; CYP71AV1, amorpha-4; 11-diene monooxygenase; ADH1, artemisinic alcohol dehydrogenase; ALDH1, artemisinic aldehyde dehydrogenase 1; Dbr2, artemisinic aldehyde Δ11 (13) reductase; CPR1, cognate reductase of CYP71AV1. Arrows with frames showed the gene elements manipulated by Keasling’s team for artemisinic acid production engineering strain construction.
Although widespread investigations have been carried out, the mechanism of action of artemisinin is still incompletely clarified. It has been widely accepted that the anti-malarial activity of artemisinin is largely dependent on the unusual endoperoxide since derivatives lacking the endoperoxide bridge are discovered to be devoid of antimalarial activity, and the activity could be enhanced by high oxygen tension and by the addition of other free-radical-generating compounds, while some radical scavengers could block the antimalarial activity [53]. Considerable evidence has proven that the killing parasite’s ability of artemisinin-based combination therapies is mediated by free radicals, which are produced from the endoperoxide bridge [54]. The degradation of the endoperoxide bridge in a heme-dependent process could form carbon-centered radicals, which then alkylate multiple targets including heme and proteins at the pathogenic Plasmodium blood stage and lead to the conversion of heme to hemozoin and finally lead to the death of the parasite [55].
3.2. Key Enzymes Involved in The Biosynthesis Pathway of Artemisinin
The large demand for artemisinin-based combination therapies has caused artemisinin to fall in short supply. To provide more alternative sources, the biosynthesis pathway of artemisinin has been investigated for many years and remarkable achievements have been obtained. Like other regular sesquiterpenes, artemisinin’s biosynthesis precursor is farnesyl pyrophosphate (FPP), which is formed by the condensation of three IPP molecules by either the MVA or plastid non-MVA pathway, respectively [56]. To verify the origin of the precursors, a plant of A. annua was grown in an atmosphere containing labeled 13CO2 for 100 min. Following a chase period of 10 days, artemisinin was isolated and analyzed by 13C NMR spectroscopy. The result shows that the precursor IPP can be provided by both the MVA pathway and the non-MVA pathway. As shown in Figure 2, DMAPP was initially provided by MVA origin and then transferred to the plastid, where an IPP unit of non-MVA origin is used for elongation to form geranyl diphosphate (GPP). In the subsequent step, GPP is exported to the cytosolic compartment and converted into FPP using IPP from the MVA pathway [57] (Figure 2). After FPP is formed, the first committed step of artemisinin biosynthesis is the conversion of FPP to amorphadiene by the terpene synthase enzyme amorphadiene synthase (ADS). To explore the catalysis mechanism of ADS, deuterium-labeled FPP at H-1 position was used as the substrate to trace the H-1 hydrogen migration of FPP during cyclization. 1H NMR results of amorphadiene showed that one of the hydrogen Ha-1 of FPP migrated to H-10 of amorphadiene, while the other hydrogen Hb-1 remained at its position to label amorphadiene H-6. These observations indicated that ADS may act through an initial formation of a bisabolyl cation intermediate through 1,6-ring closure and one 1,3-hydride shift. Bisabolyl carbocation intermediate would then undergo hydride shift through one direct suprafacial 1,3-shift of axial Ha-1 to C-7 (Figure 2), resulting in the correct cis-decalin configuration at C-1 and C-6 of amorphadiene [58][59][60][61]. Following the formation of amorpha-4,11-diene, a cytochrome P450, CYP71AV1, was cloned from A. annua and characterized by expression in Saccharomyces cerevisiae. CYP71AV1 could catalyze the multiple oxidation steps of amorpha-4,11-diene to produce artemisinic alcohol and artemisinic aldehyde, and finally yield artemisinic acid [62]. In addition, two genes encoding putative artemisinic alcohol dehydrogenase (ADH1) and artemisinic aldehyde dehydrogenase 1 (ALDH1) were characterized from A. annua glandular trichomes [63]. ADH1 is a NAD-dependent alcohol dehydrogenase of the medium-chain dehydrogenase/reductase superfamily, with specificity towards artemisinic alcohol. ALDH1 could effectively convert artemisinic aldehyde to artemisinic acid [64].
It is obvious that the Δ11 (13) double bond in amorpha-4,11-diene is reduced during the biosynthesis of artemisinin, which is assumed to occur in artemisinic aldehyde. A corresponding gene, Dbr2, was cloned and characterized from A. annua [65]. It could specifically reduce artemisinic aldehyde to produce dihydroartemisinic aldehyde, which could be then converted to dihydroartemisinic acid by ALDH1. Further study showed that ALDH1 could also catalyze the oxidation of artemisinic aldehyde as CYP71AV1 did [31]. Conversely, CYP71AV1 cannot catalyze the oxidation of dihydroartemisinic aldehyde. Meanwhile, experimental results showed that there was no direct enzymatic conversion of artemisinic acid into dihydroartemisinic acid. Therefore, there should be two branches that exist during artemisinin biosynthesis [66]. It is well accepted that the primary route is through dihydroartemisinic acid, and the route through artemisinic acid is a side pathway [67][68][69]. From dihydroartemisinic acid, biosynthesis of artemisinin still requires a photooxidative formation of the endoperoxide ring. However, the details of this process, such as the potential involvement of additional enzyme activities, are currently unclear. In 2004, there was a report that, through using the cell-free extracts of A. annua, realized the bioconversion of artemisinic acid to artemisinin, but the activity was not observed when using artemisinic acid as the only substrate [70]. Thus, the enzyme in charge of this reaction is still a question. One possibility is that artemisinic acid could be converted into several other compounds such as arteannuin B non-enzymatically, which is later transformed into artemisinin [71]. Another possibility is that dihydroartemisinic acid could undergo rapid plant pigment photosensitized oxidation, followed by subsequent spontaneous oxidation to form artemisinin [72].
3.3. Microbial Production of Artemisinic Acid
On the basis of the biosynthesis pathway elucidation, increasingly more attention on artemisinin is now shifting to its microbial production. Particularly represented by Dr. Jay D. Keasling, his team has made great achievements in this field [73]. They combined the biological synthesis of the earlier steps to produce the precursor artemisinic acid and the organic synthetic steps of artemisinic acid to produce artemisinin together and realized the industrial production of semi-synthetic artemisinin for commerce needs. They first constructed the biosynthesis pathway of amorphadiene in E. coli. Compared with the expression of DXP pathway genes, a dramatic increase in isoprenoid precursor production was observed when the S. cerevisiae MVA pathway was heterologously expressed in E. coli. Thus, two plasmids were correspondingly designed. One encoded the MevT operon (known as the ‘top pathway’), which comprises three genes (atoB, ERG13, and tHMG1) that are needed for the conversion of acetyl-CoA to MVA. The second plasmid encoded the MevB operon (known as the ‘bottom pathway’) comprising five genes (idi, ispA, MVD1, ERG8, and ERG12) for the conversion of MVA to FPP. These two plasmids were subsequently expressed in E. coli with the codon-optimized amorphadiene synthase (ADS) gene together. Combined with the optimization of the fermentation conditions, the production of amorphadiene could reach 0.5 g per liter in E. coli [74][75][76]. Following this is the next stage: after the identification of CYP71AV1, this project meets a quandary that although the amorphadiene was produced with a higher yield in E. coli than in S. cerevisiae, E. coli is unsuitable for the expression of the P450 enzyme CYP71AV1, which is crucial for the following steps. Thus, in this stage, Keasling’s team switched the expression system of artemisinin to S. cerevisiae. Following this, a series of gene manipulations were performed, including: (1) The S. cerevisiae strain was engineered to overexpress the MVA pathway, and all genes were integrated into the genome; (2) ADS and CYP71AV1 genes were constructed as plasmid borne; (3) overexpression of a 3-hydroxy-3-methylglutaryl-CoA reductase (tHMGR) occurred to improve the production of amorphadiene; (4) downregulation of ERG9 occurred, which encodes squalene synthase, catalyzing the first step in the sterol biosynthetic pathway to inhibit the flux from FPP to sterol; (5) a methionine repressible promoter PMET3 was used to increase amorphadiene production; (6) the ADS gene was expressed under the control of the GAL1 promoter; (7) the CYP71AV1 gene was expressed along with its cognate reductase (CPR1); (8) yeast strain CEN.PK2 was chosen as the host, which is capable of sporulating sufficiently; (9) every enzyme of the MVA pathway including ERG20 (the final step for the production of FPP) was overexpressed in CEN.PK2 in an effort to increase the production of amorphadiene; (10) the GAL80 gene was deleted to ensure constitutive expression of the overexpressed MVA pathway enzymes and the A. annua-derived genes; (11) the much cheaper glucose was used as the carbon source instead of galactose; (12) another two enzymes, aldehyde dehydrogenase (ALDH1) and artemisinic alcohol dehydrogenase (ADH1), were combinedly expressed with CYP71AV1, which resulted in the highest production yield of artemisinic acid. With all the above manipulations coupled with the development of the fermentation process, the production of artemisinic acid in the engineering yeast strain was finally as high as 25 g per liter [63][77][78].
3.4. Chemical Conversion to Produce Artemisinin
The final stage for artemisinin chemo-enzymatic synthesis is the chemical conversion of artemisinic acid to artemisinin (Figure 2). The chemical process involves a four-step conversion that begins with the reduction of artemisinic acid to dihydroartemisinic acid. Then, the esterification of the carboxylic acid moiety will be performed to block the subsequent formation of side products. The third step is an ‘ene-type’ reaction of the C4–C5 double bond with singlet oxygen (1O2) to produce an allylic 3-hydroperoxide. Moreover, in the final step, the allylic hydroperoxide undergoes an acid-catalyzed hock fragmentation and rearrangement to afford a ring-opened keto-aldehyde enol. Trapping of this enol with 3O2 produces a vicinal hydroperoxide aldehyde, followed by a cascade reaction of acid-catalyzed cyclization that could form an endoperoxide bridge to provide artemisinin at last [63]. Finally, through the metabolic engineering of the earlier steps using multiple gene manipulations and following synthetic organic chemistry, the anti-malaria drug artemisinin production system was successfully established and effectively used for industrial production by Sanofi company as the worldwide supplement [73]. Artemisinin is by far the most successful and representative example of the perfect combination of biosynthetic pathway research and industrial production.
4. Biosynthesis Pathway Investigation of the Terpenoid Antimicrobial Agent—Oleanolic Acid
Oleanolic acid (Table 1) is a pentacyclic triterpenoid originating from a number of medicinal plants. It has desired antimicrobial activity against various bacterial pathogens and viruses [15][79][80][81][82]. Furthermore, the study on this antimicrobial agent is of importance because as a natural source product, there has been no resistance case toward oleanolic acid found yet [83]. The biosynthesis pathway of oleanolic acid has been relatively clear [84].
In plant cells, acetyl CoA generates DMAPP and IPP through the MVA pathway in the cytosol. IPP and DMAPP are isomerized into FPP under the action of farnesyl pyrophosphate synthase (FPS), and FPP is then converted into squalene under the action of squalene synthase (SQS). Squalene cyclooxygenase (SQE) then oxidizes squalene into a precursor molecule for primary sterol metabolism, 2,3-oxsqualene [85]. From this step, the different cyclizations of 2,3-oxidized squalene become a branching point between primary sterol and secondary triterpene metabolism. For the biosynthesis of plant sterols, the cyclization of 2,3-oxysqualene to the tetracyclic plant sterol precursor cycloartenol is mainly catalyzed by cycloartenol synthase (CAS) [86]. Conversely, the oleanolic acid biosynthetic pathway of the researchers' interest, 2,3-oxysqualene, is cyclized by β-amyrin synthase (BAS), which was first cloned from the medicinal plant ginseng and subsequently from a variety of other plants [86][87]. This pentacyclic carbon skeleton is assumed to be formed from (3S)-2,3-oxidosqualene folded in pre-chair–chair–chair conformation [88]. Opening of the epoxide ring followed by cation–π cyclization initially produces a tetracyclic dammarenyl cation. Following ring expansion and the formation of fifth ring, the lupenyl cation is formed [87]. Another ring expansion followed by a series of stereospecific 1,2-hydride shifts and the final abstraction of 12α proton produces β-amyrin [89] (Figure 3). The C-28 position of β-amyrin is then oxidized in three consecutive steps by a single cytochrome P450 enzyme, CYP716A12, to produce oleanolic acid. The key enzyme for this step—CYP716A12—was first identified in Medicago truncatula, and the study found that erythrodiol, oleanolic aldehyde, and oleanolic acid production were detected in the reaction solution catalyzed by this enzyme [90][91]. Thus, it is suggested that CYP716A12 is a C-28 oxidase of β-amyrin, catalyzing three sequential oxidation reactions of oleanane main chain C-28 rather than a one-step generation. The oleanolic acid biosynthetic pathway is shown in Figure 3.
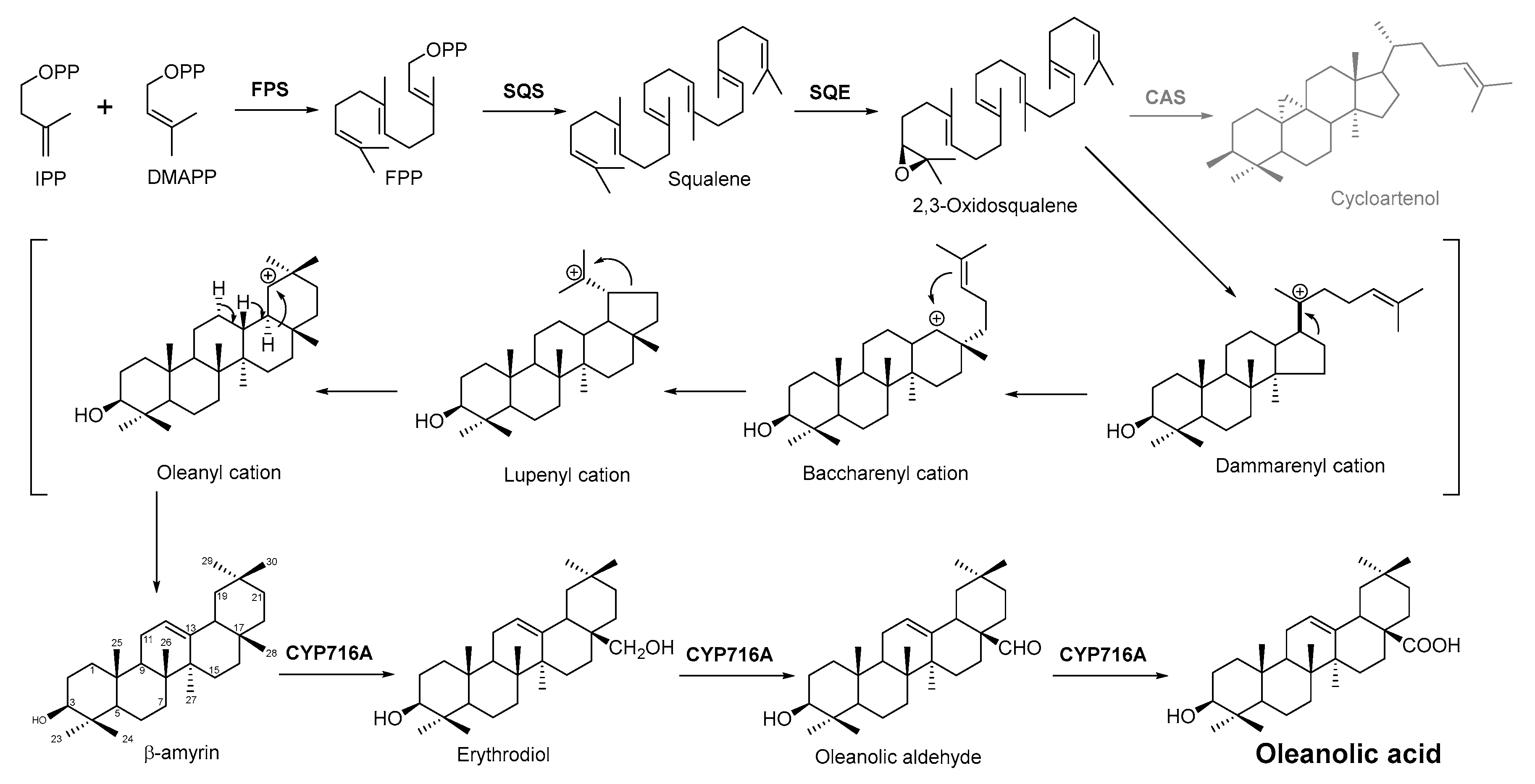
Figure 3. Biosynthesis pathway of terpenoid antimicrobial agent oleanolic acid. FPS, farnesyl diphosphate synthase; SQS, squalene synthase; SQE, squalene epoxidase; CAS, cycloartenol synthase.
With the development of synthetic biology, some conventional biosynthetic pathways were interfered with using genetic engineering to improve the target compound’s production. For example, limonene, a cyclic monoterpene of plant origin, is antimicrobially sensitive to Listeria monocytogenes and can damage its cell integrity and wall structure [92]. The most classical biosynthetic pathway of limonene is the condensation of IPP and DMAPP to form GPP by the action of geranyl pyrophosphate synthase, and limonene synthase (LS) uses GPP as a substrate to synthesize limonene. However, GPP can also subsequently condense with a molecule of IPP to form FPP, and studies have shown that the synthesis of excessive FPP hinders the efficient synthesis of monoterpenes. According to a recent report, researchers have developed an FPPS mutant (F96W, N127W; FPPSF96W, N127W) that can selectively produce GPP without further extension to FPP. In the yeast strain with high isoprene production, fppsF96W, N127W genes were combined with nine plant LS genes, and the N-terminal sequence of plasma-membrane-targeted transport peptide (TLS) was truncated. The best effect of 15.5 mg L−1 limonene on Citrus lemon tls1 (cltls1) was achieved. Moreover, an orthogonal engineering pathway was constructed. In this pathway, limonene could be produced through the condensation of IPP and DMAPP by neryl pyrophosphate (NPP) synthase to form NPP, and limonene synthase can also use NPP as a substrate to synthesize limonene. The expression of Solanum lycopersicum nerolidyl diphosphate synthase (SlNDPS1) and Citrus limon tLS2 (CltLS2) genes in the same yeast strain made the limonene yield higher than that of traditional methods (28.9 mg L−1). Under the action of glucose-induced promoter HXT1, the production of limonene can be increased to more than 900 mg L−1 by extensive pathway engineering using the FPPS competitive gene [93].
This entry is adapted from the peer-reviewed paper 10.3390/antibiotics11101380
References
- Nazzaro, F.; Fratianni, F.; De Martino, L.; Coppola, R.; De Feo, V. Effect of essential oils on pathogenic bacteria. Pharmaceuticals 2013, 6, 1451–1474.
- Burt, S.A.; Reinders, R.D. Antibacterial activity of selected plant essential oils against Escherichia coli O157:H7. Lett. Appl. Microbiol. 2003, 36, 162–167.
- Mulyaningsih, S.; Sporer, F.; Zimmermann, S.; Reichling, J.; Wink, M. Synergistic properties of the terpenoids aromadendrene and 1,8-cineole from the essential oil of Eucalyptus globulus against antibiotic-susceptible and antibiotic-resistant pathogens. Phytomedicine 2010, 17, 1061–1066.
- Di Pasqua, R.; Betts, G.; Hoskins, N.; Edwards, M.; Ercolini, D.; Mauriello, G. Membrane toxicity of antimicrobial compounds from Essential oils. J. Agric. Food Chem. 2007, 55, 4863–4870.
- Sharma, A.; Biharee, A.; Kumar, A.; Jaitak, V. Antimicrobial terpenoids as a potential substitute in overcoming antimicrobial resistance. Curr. Drug. Targets 2020, 21, 1476–1494.
- Mulat, M.; Pandita, A.; Khan, F. Medicinal plant compounds for combating the multi-drug resistant pathogenic bacteria: A Review. Curr. Pharm. Biotechnol. 2019, 20, 183–196.
- Niu, C.; Afre, S.; Gilbert, E.S. Subinhibitory concentrations of cinnamaldehyde interfere with quorum sensing. Lett. Appl. Microbiol. 2006, 43, 489–494.
- Myszka, K.; Schmidt, M.T.; Majcher, M.; Juzwa, W.; Olkowicz, M.; Czaczyk, K. Inhibition of quorum sensing-related biofilm of Pseudomonas fluorescens KM121 by Thymus vulgare essential oil and its major bioactive compounds. Int. Biodeter. Biodegr. 2016, 114, 252–259.
- Mittal, R.P.; Rana, A.; Jaitak, V. Essential Oils: An impending substitute of synthetic antimicrobial agents to overcome antimicrobial resistance. Curr. Drug Targets 2019, 20, 605–624.
- Ahmad, A.; Khan, A.; Yousuf, S.; Khan, L.A.; Manzoor, N. Proton translocating ATPase mediated fungicidal activity of eugenol and thymol. Fitoterapia 2010, 81, 1157–1162.
- Khan, I.; Bahuguna, A.; Shukla, S.; Aziz, F.; Chauhan, A.K.; Ansari, M.B.; Bajpai, V.K.; Huh, Y.S.; Kang, S.C. Antimicrobial potential of the food-grade additive carvacrol against uropathogenic E. coli based on membrane depolarization, reactive oxygen species generation, and molecular docking analysis. Microb. Pathog. 2020, 142, 104046.
- Domadia, P.; Swarup, S.; Bhunia, A.; Sivaraman, J.; Dasgupta, D. Inhibition of bacterial cell division protein FtsZ by cinnamaldehyde. Biochem. Pharmacol. 2007, 74, 831–840.
- Naz, F.; Kumar, M.; Koley, T.; Sharma, P.; Haque, M.A.; Kapil, A.; Kumar, M.; Kaur, P.; Ethayathulla, A.S. Screening of plant-based natural compounds as an inhibitor of FtsZ from Salmonella typhi using the computational, biochemical and in vitro cell-based studies. Int. J. Biol. Macromol. 2022, 219, 428–437.
- Zhou, F.; Ji, B.; Zhang, H.; Jiang, H.; Yang, Z.; Li, J.; Li, J.; Ren, Y.; Yan, W. Synergistic effect of thymol and carvacrol combined with chelators and organic acids against Salmonella Typhimurium. J. Food Prot. 2007, 70, 1704–1709.
- Fontanay, S.; Grare, M.; Mayer, J.; Finance, C.; Duval, R.E. Ursolic, oleanolic and betulinic acids: Antibacterial spectra and selectivity indexes. J. Ethnopharmacol. 2008, 120, 272–276.
- Hikal, D.M. Antibacterial Activity of Piperine and Black Pepper Oil. Biosci. Biotechnol. Res. Asia 2018, 15, 877–880.
- Khan, I.A.; Mirza, Z.M.; Kumar, A.; Verma, V.; Qazi, G.N. Piperine, a phytochemical potentiator of ciprofloxacin against Staphylococcus aureus. Antimicrob. Agents Chemother. 2006, 50, 810–812.
- Mittal, R.P.; Jaitak, V. Plant-derived natural alkaloids as new antimicrobial and adjuvant agents in existing antimicrobial therapy. Curr. Drug Targets 2019, 20, 1409–1433.
- Su, F.; Wang, J. Berberine inhibits the MexXY-OprM efflux pump to reverse imipenem resistance in a clinical carbapenem-resistant Pseudomonas aeruginosa isolate in a planktonic state. Exp. Ther. Med. 2018, 15, 467–472.
- Laudadio, E.; Cedraro, N.; Mangiaterra, G.; Citterio, B.; Mobbili, G.; Minnelli, C.; Bizzaro, D.; Biavasco, F.; Galeazzi, R. Natural alkaloid berberine activity against Pseudomonas aeruginosa MexXY-mediated aminoglycoside resistance: In Silico and in vitro Studies. J. Nat. Prod. 2019, 82, 1935–1944.
- Mujtaba, M.A.; Akhter, M.H.; Alam, M.S.; Ali, M.D.; Hussain, A. An updated review on therapeutic potential and recent advances in drug delivery of berberine: Current status and future prospect. Curr. Pharm. Biotechnol. 2022, 23, 60–71.
- Wei, W.Y.; Du, H.X.; Shao, C.Y.; Zhou, H.F.; Lu, Y.Y.; Yu, L.; Wan, H.T.; He, Y. Screening of antiviral components of ma huang tang and investigation on the ephedra alkaloids efficacy on influenza virus type A. Front. Pharmacol. 2019, 10, 961.
- He, N.; Wang, P.; Wang, P.; Ma, C.; Kang, W. Antibacterial mechanism of chelerythrine isolated from root of Toddalia asiatica (Linn) Lam. BMC Complement. Altern. Med. 2018, 18, 261.
- Prachayasittikul, V.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. 8-Hydroxyquinolines: A review of their metal chelating properties and medicinal applications. Drug Des. Dev. Ther. 2013, 7, 1157–1178.
- Houdkova, M.; Rondevaldova, J.; Doskocil, I.; Kokoska, L. Evaluation of antibacterial potential and toxicity of plant volatile compounds using new broth microdilution volatilization method and modified MTT assay. Fitoterapia 2017, 118, 56–62.
- McMahon, J.B.; Currens, M.J.; Gulakowski, R.J.; Buckheit, R.W.; Lackman-Smith, C.; Hallock, Y.F.; Boyd, M.R. Michellamine B, a novel plant alkaloid, inhibits human immunodeficiency virus-induced cell killing by at least two distinct mechanisms. Antimicrob. Agents Chemother. 1995, 39, 484–488.
- Hamoud, R.; Reichling, J.; Wink, M. Synergistic antibacterial activity of the combination of the alkaloid sanguinarine with EDTA and the antibiotic streptomycin against multidrug resistant bacteria. J. Pharm. Pharmacol. 2015, 67, 264–273.
- Beuria, T.K.; Santra, M.K.; Panda, D. Sanguinarine blocks cytokinesis in bacteria by inhibiting FtsZ assembly and bundling. Biochemistry 2005, 44, 16584–16593.
- Yin, S.J.; Rao, G.X.; Wang, J.; Luo, L.Y.; He, G.H.; Wang, C.Y.; Ma, C.Y.; Luo, X.X.; Hou, Z.; Xu, G.L. Roemerine improves the survival rate of septicemic BALB/c mice by increasing the cell membrane permeability of Staphylococcus aureus. PLoS ONE 2015, 10, e0143863.
- Avci, F.G.; Atas, B.; Aksoy, C.S.; Kurpejovic, E.; Toplan, G.G.; Gurer, C.; Guillerminet, M.; Orelle, C.; Jault, J.M.; Akbulut, B.S. Repurposing bioactive aporphine alkaloids as efflux pump inhibitors. Fitoterapia 2019, 139, 104371.
- Costa, R.S.; Lins, M.O.; Le Hyaric, M.; Barros, T.F.; Velozo, E.S. In vitro antibacterial effects of Zanthoxylum tingoassuiba root bark extracts and two of its alkaloids against multiresistant Staphylococcus aureus. Rev. Bras. Farmacogn. 2017, 27, 195–198.
- Guzman, J.D.; Wube, A.; Evangelopoulos, D.; Gupta, A.; Hufner, A.; Basavannacharya, C.; Rahman, M.M.; Thomaschitz, C.; Bauer, R.; McHugh, T.D.; et al. Interaction of N-methyl-2-alkenyl-4-quinolones with ATP-dependent MurE ligase of Mycobacterium tuberculosis: Antibacterial activity, molecular docking and inhibition kinetics. J. Antimicrob. Chemother. 2011, 66, 1766–1772.
- Hochfellner, C.; Evangelopoulos, D.; Zloh, M.; Wube, A.; Guzman, J.D.; McHugh, T.D.; Kunert, O.; Bhakta, S.; Bucar, F. Antagonistic effects of indoloquinazoline alkaloids on antimycobacterial activity of evocarpine. J. Appl. Microbiol. 2015, 118, 864–872.
- Celiz, G.; Daz, M.; Audisio, M.C. Antibacterial activity of naringin derivatives against pathogenic strains. J. Appl. Microbiol. 2011, 111, 731–738.
- Pyrzynska, K. Hesperidin: A Review on Extraction Methods, Stability and Biological Activities. Nutrients 2022, 14, 2387.
- Babu, K.S.; Babu, T.H.; Srinivas, P.V.; Sastry, B.S.; Kishore, K.H.; Murty, U.S.; Rao, J.M. Synthesis and in vitro study of novel 7-O-acyl derivatives of Oroxylin A as antibacterial agents. Bioorg. Med. Chem. Lett. 2005, 15, 3953–3956.
- Liu, R.; Zhao, B.; Wang, D.E.; Yao, T.Y.; Pang, L.; Tu, Q.; Ahmed, S.M.; Liu, J.J.; Wang, J.Y. Nitrogen-containing apigenin analogs: Preparation and biological activity. Molecules 2012, 17, 14748–14764.
- Chinnam, N.; Dadi, P.K.; Sabri, S.A.; Ahmad, M.; Kabir, M.A.; Ahmad, Z. Dietary bioflavonoids inhibit Escherichia coli ATP synthase in a differential manner. Int. J. Biol. Macromol. 2010, 46, 478–486.
- Navarro-Martinez, M.D.; Navarro-Peran, E.; Cabezas-Herrera, J.; Ruiz-Gomez, J.; Garcia-Canovas, F.; Rodriguez-Lopez, J.N. Antifolate activity of epigallocatechin gallate against Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 2005, 49, 2914–2920.
- Pejin, B.; Ciric, A.; Markovic, J.D.; Glamoclija, J.; Nikolic, M.; Stanimirovic, B.; Sokovic, M. Quercetin potently reduces biofilm formation of the strain Pseudomonas aeruginosa PAO1 in vitro. Curr. Pharm. Biotechnol. 2015, 16, 733–737.
- Cushnie, T.P.; Lamb, A.J. Detection of galangin-induced cytoplasmic membrane damage in Staphylococcus aureus by measuring potassium loss. J. Ethnopharmacol. 2005, 101, 243–248.
- Gradisar, H.; Pristovsek, P.; Plaper, A.; Jerala, R. Green tea catechins inhibit bacterial DNA gyrase by interaction with its ATP binding site. J. Med. Chem. 2007, 50, 264–271.
- Fathima, A.; Rao, J.R. Selective toxicity of Catechin-a natural flavonoid towards bacteria. Appl. Microbiol. Biotechnol. 2016, 100, 6395–6402.
- Zhao, Q.; Chen, X.Y.; Martin, C. Scutellaria baicalensis, the golden herb from the garden of Chinese medicinal plants. Sci. Bull. 2016, 61, 1391–1398.
- Wang, J.; Zhu, J.; Meng, J.; Qiu, T.; Wang, W.; Wang, R.; Liu, J. Baicalin inhibits biofilm formation by influencing primary adhesion and aggregation phases in Staphylococcus saprophyticus. Vet. Microbiol. 2021, 262, 109242.
- Liu, N.; Zhang, N.; Zhang, S.; Zhang, L.; Liu, Q. Phloretin inhibited the pathogenicity and virulence factors against Candida albicans. Bioengineered 2021, 12, 2420–2431.
- Wang, D.; Xie, K.; Zou, D.; Meng, M.; Xie, M. Inhibitory effects of silybin on the efflux pump of methicillin-resistant Staphylococcus aureus. Mol. Med. Rep. 2018, 18, 827–833.
- Yin, Z.; Dickschat, J.S. Engineering fungal terpene biosynthesis. Nat. Prod. Rep. 2022. Advance Article.
- Bergman, M.E.; Davis, B.; Phillips, M.A. Medically useful plant terpenoids: Biosynthesis, occurrence, and mechanism of action. Molecules 2019, 24, 3961.
- Saha, P.; Rahman, F.I.; Hussain, F.; Rahman, S.M.A.; Rahman, M.M. Antimicrobial diterpenes: Recent development from natural sources. Front. Pharmacol. 2021, 12, 820312.
- Tu, Y. The discovery of artemisinin (qinghaosu) and gifts from Chinese medicine. Nat. Med. 2011, 17, 1217–1220.
- Tu, Y. Artemisinin-a gift from traditional Chinese medicine to the world (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2016, 55, 10210–10226.
- O’Neill, P.M.; Barton, V.E.; Ward, S.A. The molecular mechanism of action of artemisinin—The debate continues. Molecules 2010, 15, 1705–1721.
- Meshnick, S.R.; Taylor, T.E.; Kamchonwongpaisan, S. Artemisinin and the antimalarial endoperoxides: From herbal remedy to targeted chemotherapy. Microbiol. Rev. 1996, 60, 301–315.
- Klonis, N.; Crespo-Ortiz, M.P.; Bottova, I.; Abu-Bakar, N.; Kenny, S.; Rosenthal, P.J.; Tilley, L. Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion. Proc. Natl. Acad. Sci. USA 2011, 108, 11405–11410.
- Wen, W.; Yu, R. Artemisinin biosynthesis and its regulatory enzymes: Progress and perspective. Pharmacogn. Rev. 2011, 5, 189–194.
- Schramek, N.; Wang, H.; Römisch-Margl, W.; Keil, B.; Radykewicz, T.; Winzenhörlein, B.; Beerhues, L.; Bacher, A.; Rohdich, F.; Gershenzon, J.; et al. Artemisinin biosynthesis in growing plants of Artemisia annua. A 13CO2 study. Phytochemistry 2010, 71, 179–187.
- Bouwmeester, H.J.; Wallaart, T.E.; Janssen, M.H.; van Loo, B.; Jansen, B.J.; Posthumus, M.A.; Schmidt, C.O.; De Kraker, J.W.; König, W.A.; Franssen, M.C. Amorpha-4,11-diene synthase catalyses the first probable step in artemisinin biosynthesis. Phytochemistry 1999, 52, 843–854.
- Mercke, P.; Bengtsson, M.; Bouwmeester, H.J.; Posthumus, M.A.; Brodelius, P.E. Molecular cloning, expression, and characterization of amorpha-4,11-diene synthase, a key enzyme of artemisinin biosynthesis in Artemisia annua L. Arch. Biochem. Biophys. 2000, 381, 173–180.
- Picaud, S.; Mercke, P.; He, X.; Sterner, O.; Brodelius, M.; Cane, D.E.; Brodelius, P.E. Amorpha-4,11-diene synthase: Mechanism and stereochemistry of the enzymatic cyclization of farnesyl diphosphate. Arch. Biochem. Biophys. 2006, 448, 150–155.
- Kim, S.H.; Heo, K.; Chang, Y.J.; Park, S.H.; Rhee, S.K.; Kim, S.U. Cyclization mechanism of amorpha-4,11-diene synthase, a key enzyme in artemisinin biosynthesis. J. Nat. Prod. 2006, 69, 758–762.
- Teoh, K.H.; Polichuk, D.R.; Reed, D.W.; Nowak, G.; Covello, P.S. Artemisia annua L. (Asteraceae) trichome-specific cDNAs reveal CYP71AV1, a cytochrome P450 with a key role in the biosynthesis of the antimalarial sesquiterpene lactone artemisinin. FEBS Lett. 2006, 580, 1411–1416.
- Paddon, C.J.; Westfall, P.J.; Pitera, D.J.; Benjamin, K.; Fisher, K.; McPhee, D.; Leavell, M.D.; Tai, A.; Main, A.; Eng, D.; et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 2013, 496, 528–532.
- Teoh, K.; Polichuk, D.; Reed, D.; Covello, P. Molecular cloning of an aldehyde dehydrogenase implicated in artemisinin biosynthesis in Artemisia annua. Botany 2009, 87, 635–642.
- Zhang, Y.; Teoh, K.H.; Reed, D.W.; Maes, L.; Goossens, A.; Olson, D.J.; Ross, A.R.; Covello, P.S. The molecular cloning of artemisinic aldehyde Δ11(13) reductase and its role in glandular trichome-dependent biosynthesis of artemisinin in Artemisia annua. J. Biol. Chem. 2008, 283, 21501–21508.
- Bertea, C.M.; Freije, J.R.; van der Woude, H.; Verstappen, F.W.; Perk, L.; Marquez, V.; De Kraker, J.W.; Posthumus, M.A.; Jansen, B.J.; de Groot, A.; et al. Identification of intermediates and enzymes involved in the early steps of artemisinin biosynthesis in Artemisia annua. Planta Med. 2005, 71, 40–47.
- Wallaart, T.E.; Pras, N.; Quax, W.J. Isolation and identification of dihydroartemisinic acid hydroperoxide from Artemisia annua: A novel biosynthetic precursor of artemisinin. J. Nat. Prod. 1999, 62, 1160–1162.
- Sy, L.-K.; Brown, G.D. The mechanism of the spontaneous autoxidation of dihydroartemisinic acid. Tetrahedron 2002, 58, 897–908.
- Arsenault, P.R.; Wobbe, K.K.; Weathers, P.J. Recent advances in artemisinin production through heterologous expression. Curr. Med. Chem. 2008, 15, 2886–2896.
- Zhang, Y.S.; Liu, B.Y.; Li, Z.Q.; Ye, H.C.; Wang, H.; Li, G.F.; Han, J.L. Molecular cloning of a classical plant peroxidase from Artemisia annua and its effect on the biosynthesis of artemisinin in vitro. Acta Bot. Sin. 2004, 46, 1338–1346.
- Brown, G.D.; Sy, L.-K. In vivo transformations of artemisinic acid in Artemisia annua plants. Tetrahedron 2007, 63, 9548–9566.
- Brown, G.D.; Sy, L.-K. In vivo transformations of dihydroartemisinic acid in Artemisia annua plants. Tetrahedron 2004, 60, 1139–1159.
- Paddon, C.J.; Keasling, J.D. Semi-synthetic artemisinin: A model for the use of synthetic biology in pharmaceutical development. Nat. Rev. Microbiol. 2014, 12, 355–367.
- Martin, V.J.J.; Pitera, D.J.; Withers, S.T.; Newman, J.D.; Keasling, J.D. Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nat. Biotechnol. 2003, 21, 796–802.
- Martin, V.J.; Yoshikuni, Y.; Keasling, J.D. The in vivo synthesis of plant sesquiterpenes by Escherichia coli. Biotechnol. Bioeng. 2001, 75, 497–503.
- Newman, J.D.; Marshall, J.; Chang, M.; Nowroozi, F.; Paradise, E.; Pitera, D.; Newman, K.L.; Keasling, J.D. High-level production of amorpha-4,11-diene in a two-phase partitioning bioreactor of metabolically engineered Escherichia coli. Biotechnol. Bioeng. 2006, 95, 684–691.
- Chang, M.C.; Keasling, J.D. Production of isoprenoid pharmaceuticals by engineered microbes. Nat. Chem. Biol. 2006, 2, 674–681.
- Ro, D.K.; Paradise, E.M.; Ouellet, M.; Fisher, K.J.; Newman, K.L.; Ndungu, J.M.; Ho, K.A.; Eachus, R.A.; Ham, T.S.; Kirby, J.; et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 2006, 440, 940–943.
- Jimenez-Arellanes, A.; Meckes, M.; Torres, J.; Luna-Herrera, J. Antimycobacterial triterpenoids from Lantana hispida (Verbenaceae). J. Ethnopharmacol. 2007, 111, 202–205.
- Kozai, K.; Suzuki, J.; Okada, M.; Nagasaka, N. Effect of oleanolic acid-cyclodextrin inclusion compounds on dental caries by in vitro experiment and rat-caries model. Microbios 1999, 97, 179–188.
- Horiuchi, K.; Shiota, S.; Hatano, T.; Yoshida, T.; Kuroda, T.; Tsuchiya, T. Antimicrobial activity of oleanolic acid from Salvia officinalis and related compounds on vancomycin-resistant enterococci (VRE). Biol. Pharm. Bull. 2007, 30, 1147–1149.
- Castellano, J.M.; Ramos-Romero, S.; Perona, J.S. Oleanolic Acid: Extraction, Characterization and Biological Activity. Nutrients 2022, 14, 623.
- Wolska, K.; Grudniak, A.M.; Fiecek, B.; Kraczkiewicz-Dowjat, A.; Kurek, A. Antibacterial activity of oleanolic and ursolic acids and their derivatives. Open Life Sci. 2010, 5, 543–553.
- Pollier, J.; Goossens, A. Oleanolic acid. Phytochemistry 2012, 77, 10–15.
- Phillips, D.R.; Rasbery, J.M.; Bartel, B.; Matsuda, S.P. Biosynthetic diversity in plant triterpene cyclization. Curr. Opin. Plant Biol. 2006, 9, 305–314.
- Abe, I. Enzymatic synthesis of cyclic triterpenes. Nat. Prod. Rep. 2007, 24, 1311–1331.
- Kushiro, T.; Shibuya, M.; Ebizuka, Y. β-amyrin synthase—Cloning of oxidosqualene cyclase that catalyzes the formation of the most popular triterpene among higher plants. Eur. J. Biochem. 1998, 256, 238–244.
- Abe, I.; Rohmer, M.; Prestwich, G.D. Enzymatic Cyclization of Squalene and Oxidosqualene to Sterols and Triterpenes. Chem. Rev. 1993, 93, 2189–2206.
- Seo, S.; Yoshimura, Y.; Uomori, A.; Takeda, K.; Seto, H.; Ebizuka, Y.; Sankawa, U. Biosynthesis of triterpenes, ursolic acid and oleanolic acid in tissue cultures of Rabdosia japonica Hara fed mevalonolactone and acetate. J. Am. Chem. Soc. 1988, 110, 1740–1745.
- Fukushima, E.O.; Seki, H.; Ohyama, K.; Ono, E.; Umemoto, N.; Mizutani, M.; Saito, K.; Muranaka, T. CYP716A subfamily members are multifunctional oxidases in triterpenoid biosynthesis. Plant Cell Physiol. 2011, 52, 2050–2061.
- Carelli, M.; Biazzi, E.; Panara, F.; Tava, A.; Scaramelli, L.; Porceddu, A.; Graham, N.; Odoardi, M.; Piano, E.; Arcioni, S.; et al. Medicago truncatula CYP716A12 is a multifunctional oxidase involved in the biosynthesis of hemolytic saponins. Plant Cell 2011, 23, 3070–3081.
- Han, Y.; Sun, Z.; Chen, W. Antimicrobial Susceptibility and Antibacterial Mechanism of Limonene against Listeria monocytogenes. Molecules 2019, 25, 33.
- Cheng, S.; Liu, X.; Jiang, G.; Wu, J.; Zhang, J.L.; Lei, D.; Yuan, Y.J.; Qiao, J.; Zhao, G.R. Orthogonal Engineering of Biosynthetic Pathway for Efficient Production of Limonene in Saccharomyces cerevisiae. ACS Synth. Biol. 2019, 8, 968–975.
This entry is offline, you can click here to edit this entry!