Monoclonal gammopathy of clinical significance (MGCS) refers to a recently coined term describing a complex and heterogeneous group of nonmalignant monoclonal gammopathies. These patients are characterized by the presence of a commonly small clone and the occurrence of symptoms that may be associated with the clone or with the monoclonal protein through diverse mechanisms. This is an evolving, challenging, and rapidly changing field. Patients are classified according to the key organ or system involved, with kidneys, skin, nerves, and eyes being the most frequently affected. However, multiorgan involvement may be the most relevant clinical feature at the presentation or during the course.
- monoclonal gammopathy of clinical significance
- diagnosis
- prognosis
- treatment
1. Introduction
| Monoclonal Gammopathies |
|---|
| ▪ Cold agglutinin disease * |
| ▪ IgM monoclonal gammopathy of undetermined significance |
| ▪ Non-IgM monoclonal gammopathy of undetermined significance |
| ▪ Monoclonal gammopathy of renal significance * |
| Diseases with monoclonal immunoglobulin deposition |
| ▪ Immunoglobulin-related (AL) amyloidosis |
| ▪ Monoclonal immunoglobulin deposition disease |
| Heavy chain diseases |
| ▪ Mu heavy chain disease |
| ▪ Gamma heavy chain disease |
| ▪ Alpha heavy chain disease |
| Plasma cell neoplasms |
| ▪ Plasmacytoma |
| ▪ Plasma cell myeloma |
| ▪ Plasma cell neoplasms with associated paraneoplastic syndrome |
| ◊ POEMS |
| ◊ TEMPI |
| ◊ AESOP * |
| IgM Monoclonal Gammopathy of Undetermined Significance (MGUS) |
|---|
| ▪ IgM MGUS, plasma cell type * |
| ▪ IgM MGUS, NOS * |
| Primary cold agglutinin disease * |
| Heavy chain diseases |
| ▪ Mu heavy chain disease |
| ▪ Gamma heavy chain disease |
| ▪ Alpha heavy chain disease |
| Plasma cell neoplasms |
| ▪ Non-IgM monoclonal gammopathy of undetermined significance |
| ▪ Multiple myeloma (Plasma cell myeloma) * |
| ◊ Multiple myeloma NOS |
| ◊ Multiple myeloma with recurrent genetic abnormality |
| - Multiple myeloma with CCND family translocation |
| - Multiple myeloma with MAF family translocation |
| - Multiple myeloma with NSD2 translocation |
| - Multiple myeloma with hyperdiploidy |
| ▪ Solitary plasmacytoma of bone |
| ▪ Extraosseous plasmacytoma |
| Monoclonal immunoglobulin deposition diseases |
| ▪ Immunoglobulin light chain amyloidosis (AL) * |
| ▪ Localized AL amyloidosis * |
| ▪ Light chain and heavy chain deposition disease |
2. Definitions
2.1. MGUS
2.1.1. Non-Ig M MGUS
2.1.2. Ig M MGUS
2.1.3. LC MGUS
2.2. MGRS
2.3. MGCS
3. Historic Note
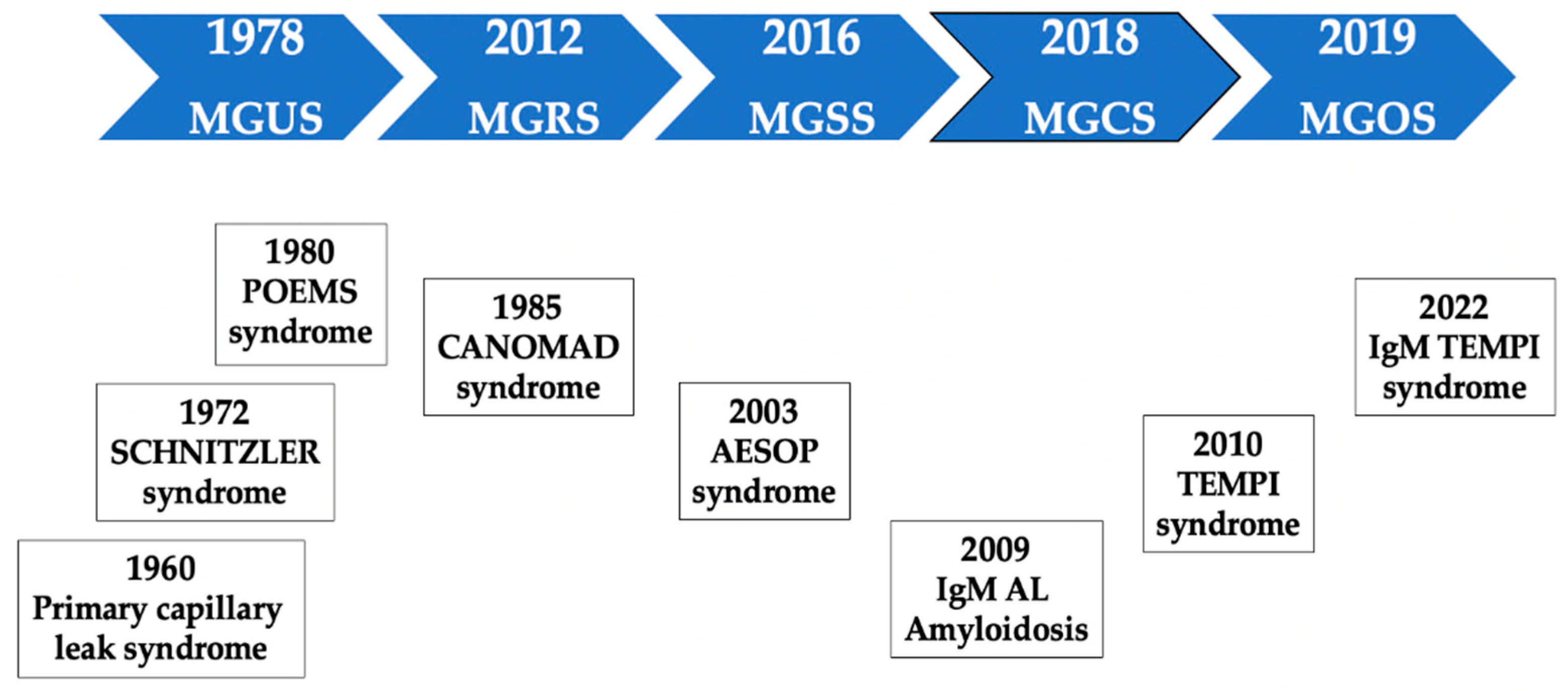
4. A Practical Classification of MGCS
5. Monoclonal Gammopathies of Renal Significance
Kidney biopsy plays a central role in this diagnosis [27][28][29]. Strikingly, in a study of 6300 patients with MG, only 160 (2.5%) had undergone a kidney biopsy. Of the 160 patients, 64 (40%) had an MGRS lesion, with AL amyloidosis the most common finding, accounting for 43.8% of these lesions. The decision to perform a kidney biopsy depended on age, the level of proteinuria, and renal function. Despite some limitations, it showed that the likelihood of performing a kidney biopsy was higher in younger patients with more severe kidney disease in terms of proteinuria and serum creatinine. The probability of reaching a diagnosis of MGRS was associated with the presence of significant proteinuria (≥1.5 g/day), hematuria and an abnormal FLCr. Therefore, a kidney biopsy should be particularly considered in patients with these characteristics [30].
The list of MGRS-associated kidney disorders is still expanding. These conditions can manifest as glomerular diseases, tubulopathies, and vascular involvement, with varying clinical presentations. Therefore, diagnosis is often challenging due to the heterogeneous presentation, the difficulty in establishing a pathogenic link between the presence of the M-protein or serum FLC and kidney disease, and the decision to perform a kidney biopsy. The high incidence of MGUS and other kidney disorders in elderly patients makes the diagnostic process even more difficult. Treatment can potentially reverse kidney disease; hence, early diagnosis is of great value [31][32]. A combined hematologic and nephrologic approach is crucial to establishing the causative role of the M-protein in the pathogenesis of kidney disease. Regarding the risk of progression to MM, MGRS had a significantly higher risk than MGUS (18% vs. 3%; p < 0.001), and this risk was 10% vs. 1% within the first year after diagnosis [33].
6. Monoclonal Gammopathies of Skin Significance
Some dermatologic entities are strongly associated with the presence of a MG, and they should be referred to as MGSS. Again, the demonstration of the association between the M-protein or the clone itself with skin damage is key. As expected, a skin biopsy plays a critical role in the diagnostic process. The direct toxicity of M-protein, host immune abnormalities, specific cytokines, and PC infiltration can, among other mechanisms, produce severe skin manifestations.
The presence of unexplained skin lesions in a patient with MG should be assessed primarily by a hematologist and a dermatologist, with the participation of other specialties, such as rheumatology, internal medicine, and ophthalmology, in certain situations. Every patient with MG and a new skin lesion of unknown origin should be investigated. In these cases, a skin biopsy and bone marrow examination, in addition to the standard laboratory evaluation of PC dyscrasia, should be performed [34]. Only two of these disorders will be described to emphasize the complex clinical presentation as a syndrome and the need to fulfil the diagnostic criteria.
POEMS is a rare paraneoplastic syndrome associated with a PCN [1]. Its acronym stands for polyradiculoneuropathy (P), organomegaly (O), endocrinopathy (E), monoclonal gammopathy (M), and skin changes (S). The components of this pentad are not always required for diagnosis. This entity is also called osteosclerotic myeloma due to the presence of osteosclerotic bone lesions in most patients. Other features may include lymphadenopathy, and in these patients, a PC variant of Castleman disease may be present.
7. Monoclonal Gammopathies of Ocular Significance
The cornea is normally a transparent structure. Several abnormalities can cause corneal opacities, making vision difficult. Patients with MG should be included in the differential diagnosis of acquired corneal opacities, as this ocular finding could be the initial manifestation of a systemic disease that can potentially be life threatening. MGOS is a rare subset of MGCS that occurs secondary to PC disorders and causes ocular manifestations. The most frequent ocular M-protein–related condition is crystalline keratopathy consisting of Ig deposition, corneal thickening, photophobia, and finally, visual loss. In a recent series of 23 patients with paraproteinemic keratopathy, neither ocular nor hematologic treatment afforded a durable improvement in visual acuity (recurrence after a median of 11 months), despite initial responses. Further studies are required to determine the optimal strategy to treat and prevent the relapse of ocular symptoms in patients with paraproteinemic keratopathy [35].
8. Monoclonal Gammopathies of Neurological Significance
9. MGCS as a Global and Unifying Concept
9.1. Diagnosis
| Major | Telangiectasias |
| Elevated erythropoietin and erythrocytosis | |
| Monoclonal gammopathy | |
| Minor | Perinephric fluid |
| Intrapulmonary shunting | |
| Other | Venous thrombosis |
9.2. Treatment
10. Conclusions
This entry is adapted from the peer-reviewed paper 10.3390/cancers14215247
References
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; de Oliveira Araujo, I.B.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748.
- Campo, E.; Jaffe, E.S.; Cook, J.R.; Quintanilla-Martinez, L.; Swerdlow, S.H.; Anderson, K.C.; Brousset, P.; Cerroni, L.; DeLeval, L.; Dirnhoffer, S.; et al. The International Consensus Classification of Mature Lymphoid Neoplasms: A Report from the Clinical Advisory Committee. Blood 2022, 140, 1229–1253.
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548.
- Bladé, J. Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2006, 355, 2765–2770.
- Van de Donk, N.W.C.J.; Mutis, T.; Poddighe, P.J.; Lokhorst, H.M.; Zweegman, S. Diagnosis, risk stratification and management of monoclonal gammopathy of undetermined significance and smoldering multiple myeloma. Int. J. Lab. Hem. 2016, 38, 110–122.
- Berenson, J.R.; Anderson, K.C.; Audell, R.A.; Boccia, R.V.; Coleman, M.; Dimopoulos, M.A.; Drake, M.T.; Fonseca, R.; Harousseau, J.L.; Joshua, D.; et al. Monoclonal gammopathy of undetermined significance: A consensus statement. Br. J. Haematol. 2010, 150, 28–38.
- Mouhieddine, T.H.; Weeks, L.D.; Ghobrial, I.M. Monoclonal gammopathy of undetermined significance. Blood 2019, 133, 2484–2494.
- Seth, S.; Zanwar, S.; Vu, L.; Kapoor, P. Monoclonal Gammopathy of Undetermined Significance: Current Concepts and Future Prospects. Curr. Hematol. Malig. Rep. 2020, 15, 45–55.
- Jiménez, C.; Prieto-Conde, M.I.; García-Álvarez, M.; Alcoceba, M.; Escalante, F.; Chillón, M.C.; García de Coca, A.; Balanzategui, A.; Cantalapiedra, A.; Aguilar, C.; et al. Unraveling the heterogeneity of IgM monoclonal gammopathies: A gene mutational and gene expression study. Ann. Hematol. 2018, 97, 475–484.
- Alcoceba, M.; García-Álvarez, M.; Medina, A.; Maldonado, R.; González-Calle, V.; Chillón, M.C.; Sarasquete, M.E.; González, M.; García-Sanz, R.; Jiménez, C. MYD88 Mutations: Transforming the Landscape of IgM Monoclonal Gammopathies. Int. J. Mol. Sci. 2022, 23, 5570.
- Bagratini, T.; Markou, A.; Patseas, D.; Mavrianou-Koutsoukou, N.; Aktypi, F.; Ivy Liacos, C.; Sklirou, A.D.; Theodorakakou, F.; Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; et al. Determination of MYD88L265P mutation fraction in IgM monoclonal gammopathies. Blood Adv. 2022, 6, 189–199.
- Drandi, D.; Decruyenaere, P.; Ferrante, M.; Offner, F.; Vandesompele, J.; Ferrero, S. Nucleic Acid Biomarkers in Waldenström Macroglobulinemia and IgM-MGUS: Current Insights and Clinical Relevance. Diagnostics 2022, 12, 969.
- Dispenzieri, A.; Katzmann, J.A.; Kyle, R.A.; Larson, D.R.; Melton, L.J., III.; Colby, C.L.; Therneau, T.M.; Clark, R.; Kumar, S.K.; Bradwell, A.; et al. Prevalence and risk of progression of light-chain monoclonal gammopathy of undetermined signifi cance: A retrospective population-based cohort study. Lancet 2010, 375, 1721–1728.
- Pelzer, B.W.; Arendt, M.; Moebus, S.; Eisele, L.; Jöckel, K.H.; Dührsen, U.; Dürig, J. Light chain monoclonal gammopathy of undetermined significance is characterized by a high disappearance rate and low risk of progression on longitudinal analysis. Ann. Hematol. 2018, 97, 1463–1469.
- Leung, N.; Bridoux, F.; Hutchinson, C.A.; Nasr, S.H.; Cockwell, P.; Fermand, J.P.; Dispenzieri, A.; Song, K.W.; Kyle, R.A. Monoclonal gammopathy of renal significance (MGRS): When MGUS is no longer undetermined or insignificant. Blood 2012, 120, 4292–4295.
- Bridoux, F.; Leung, N.; Hutchinson, C.A.; Touchard, G.; Sethi, S.; Fermand, J.P.; Picken, M.M.; Herrera, G.A.; Kastritis, E.; Merlini, G. Diagnosis of monoclonal gammopathy of renal significance. Kidney Int. 2015, 87, 698–711.
- Ciocchini, M.; Arbelide, A.; Musso, C.G. Monoclonal gammopathy of renal significance (MGRS): The characteristics and significance of a new meta-entity. Int. Urol. Nephrol. 2017, 49, 2171–2175.
- Sethi, S.; Rajkumar, S.V.; D’Agati, V.D. The Complexity and Heterogeneity of Monoclonal Immunoglobulin–Associated Renal Diseases. J. Am. Soc. Nephrol. 2018, 29, 1810–1823.
- Leung, N.; Bridoux, F.; Batuman, V.; Chaidos, A.; Cockwell, P.; D’Agati, V.D.; Dispenzieri, A.; Fervenza, F.C.; Fermand, J.P.; Gibbs, S.; et al. The evaluation of monoclonal gammopathy of renal significance: A consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat. Rev. Nephrol. 2019, 15, 45–59.
- Amaador, K.; Peeters, H.; Minnema, M.C.; Nguyen, T.Q.; Dendooven, A.; Vos, J.M.I.; Croockewit, A.J.; van de Donk, N.W.C.J.; Jacobs, J.F.M.; Wetzels, J.F.M.; et al. Monoclonal gammopathy of renal significance (MGRS): Histopathologic classification, diagnostic workup, and therapeutic options. Neth. J. Med. 2019, 77, 243–254.
- Fermand, J.P.; Bridoux, F.; Dispenzieri, A.; Jaccard, A.; Kyle, R.A.; Leung, N.; Merlini, G. Monoclonal gammopathy of clinical significance: A novel concept with therapeutic implications. Blood 2018, 132, 1478–1485.
- Dispenzieri, A. Monoclonal Gammopathy of Clinical Significance. Hematol. Am. Soc. Hematol. Educ. Program 2020, 1, 380–388.
- Kyle, R.A. Monoclonal gammopathy of undetermined significance: Natural history in 241 cases. Am. J. Med. 1978, 64, 814–826.
- Landgren, O.; Kyle, R.A.; Pfeiffer, R.M.; Katzmann, J.A.; Caporaso, N.E.; Hayes, R.B.; Dispenzieri, A.; Kumar, S.; Clark, R.J.; Baris, D.; et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: A prospective study. Blood 2009, 113, 5412–5417.
- Lipsker, D. Monoclonal gammopathy of cutaneous significance: Review of a relevant concept. J. Eur. Acad. Dermatol. Venereol. 2016, 31, 45–52.
- Karakus, S.; Gottsch, J.D.; Caturegli, P.; Eghrari, A.O. Monoclonal gammopathy of “ocular” significance. Am. J. Ophthalmol. Case Rep. 2019, 15, 100471.
- Leung, N.; Bridoux, F.; Nasr, S.H. Monoclonal gammopathy of undetermined significance. N. Engl. J. Med. 2021, 384, 1931–1941.
- Jain, A.; Haynes, R.; Kothari, J.; Khera, A.; Soares, M.; Ramasamy, K. Pathophysiology and management of monoclonal gammopathy of renal significance. Blood Adv. 2019, 3, 2409–2423.
- Klank, D.; Hoffmann, M.; Porubsky, S.; Bergner, R. Histological Findings in Kidney Biopsies of Patients with Monoclonal Gammopathy—Always a Surprise. Diagnostics 2022, 12, 1912.
- Klomjit, N.; Leung, N.; Fervenza, F.; Sethi, S.; Zand, L. Rate and predictors of finding Monoclonal Gammopathy of Renal Significance (MGRS) lesions on kidney biopsy in patients with monoclonal gammopathy. J. Am. Soc. Nephrol. 2020, 31, 2400–2411.
- Alonso-Titos, J.; Martínez-Esteban, M.D.; López, V.; León, M.; Martin-Reyes, G.; Ruiz-Esteban, P.; Hernández, D. Monoclonal gammopathy of renal significance: Early diagnosis is key. Nefrología 2021, 41, 502–513.
- Khera, A.; Panitsas, F.; Djebbari, F.; Kimberger, K.; Stern, S.; Quinn, J.; Rabin, N.; Kothari, J.; Alchi, B.; Haynes, R.; et al. Long term outcomes in monoclonal gammopathy of renal significance. Br. J. Haematol. 2019, 186, 706–716.
- Steiner, N.; Göbel, G.; Suchecki, P.; Prokop, W.; Neuwirt, H.; Gunsilius, E. Monoclonal gammopathy of renal significance (MGRS) increases the risk for progression to multiple myeloma: An observational study of 2935 MGUS patients. Oncotarget 2018, 9, 2344–2356.
- Claveau, J.S.; Wetter, D.A.; Kumar, S. Cutaneous manifestations of monoclonal gammopathy. Blood Cancer J. 2022, 12, 58.
- Garderet, L.; Al Hariri, M.; Wasielica-Poslednik, J.; Munder, M.; Kormányos, K.; Pena, C.; Gozzetti, A.; Zhou, X.; Waszczuk-Gajda, A.; Rosiñol, L.; et al. Monoclonal gammopathy of ocular significance (MGOS)—A short survey of corneal manifestations and treatment outcomes. Leuk. Lymphoma 2022, 63, 984–990.
- Rajabally, Y.A. Neuropathy and paraproteins: Review of a complex association. Eur. J. Neurol. 2011, 18, 1291–1298.
- Rison, R.A.; Beydoun, S.R. Paraproteinemic neuropathy: A practical review. BMC Neurol. 2016, 16, 13.
- Bazari, H.; Attar, E.C.; Dahl, D.M.; Upport, R.N.; Colvin, R.B. Case records of the Massachusetts General Hospital. Case 23-2010. A 49-year-old man with erythrocytosis, perinephric fluid collections, and renal failure. N. Engl. J. Med. 2010, 363, 463–475.
- Sykes, D.B.; Schoyens, W.; O’Connell, C. The TEMPI Syndrome—A Novel Multisystem Disease. N. Engl. J. Med. 2011, 365, 475–477.
- Sykes, D.B.; O’Connell, C.; Schoyens, W. The TEMPI syndrome. Blood 2020, 135, 1199–1203.
- Ruan, Y.; Zhao, X.; Pan, M. Diffuse telangiectasia: A clue to the TEMPI syndrome. JAAD Case Reports 2021, 10, 99–101.
- Xu, J.; Liu, W.; Fan, F.; Zhang, B.; Zhao, F.; Hu, Y.; Sun, C. TEMPI Syndrome: Update on Clinical Features, Management, and Pathogenesis. Front. Endocrinol. 2022, 13, 886961.
- Rögnvaldsson, S.; Love, T.J.; Thorsteinsdottir, S.; Reed, E.R.; Óskarsson, J.P.; Pétursdóttir, I.; Sigurðardóttir, G.A.; Viðarsson, B.; Önundarson, P.T.; Agnarsson, B.A.; et al. Iceland screens, treats, or prevents multiple myeloma (iStopMM): A population-based screening study for monoclonal gammopathy of undetermined significance and randomized controlled trial of follow-up strategies. Blood Cancer J. 2021, 11, 94.
- El-Khoury, H.; Lee, D.J.; Alberge, J.B.; Redd, R.; Cea-Curry, C.J.; Perry, J.; Barr, H.; Murphy, C.; Sakrikar, D.; Barnidge, D.; et al. Prevalence of monoclonal gammopathies and clinical outcomes in a high-risk US population screened by mass spectrometry: A multicentre cohort study. Lancet Haematol. 2022, 9, 340–349.