Telomeric repeat-binding factor 2 is a protein that is present at telomeres throughout the cell cycle. It is also known as TERF2, TRF2, and TRBF2, and is encoded in humans by the TERF2 gene. It is a component of the shelterin nucleoprotein complex and a second negative regulator of telomere length, playing a key role in the protective activity of telomeres. It was first reported in 1997 in the lab of Titia de Lange, where a DNA sequence similar, but not identical, to TERF1 was discovered, with respect to the Myb-domain. De Lange isolated the new Myb-containing protein sequence and called it TERF2.
- cell cycle
- telomere
- telomeres
1. Structure & Domains
TERF2 has a similar structure to that of TERF1. Both proteins carry a C-terminus Myb motif and large TERF1-related dimerization domains near their N-terminus.[1] However, both proteins exist exclusively as homodimers and do not heterodimerize with each other, as proven by co-immunoprecipitation assay analysis.[1] Also, TERF2 has a basic N-terminus, differing from TERF1’s acidic N-terminus, and was found to be much more conserved, suggesting that the two proteins have distinct functions.[2]
There are 4 domain categories on the TERF2 protein that allow it to bind to both other proteins in the shelterin protein complex, and to specific types of DNA.
1.1. TERF Homology Domain
The TERF Homology Domain (TRFH) is an area that helps to promote homodimerization of TERF2 with itself. This results in the formation of a quaternary structure that is characteristic of this protein. This TRFH domain also allows TERF2 to bind to and act as a dock for many other types of proteins. The Apollo nuclease, a shelterin accessory factor, uses the TRFH domain as a dock. The recruitment of Apollo by TERF2 allows for telomeric ends formed by DNA synthesis to be processed. By doing so, the telomere ends are able to avoid ATM kinase activation through the creation of a terminal structure.[3] SLX4, which is important in DNA repair by acting as a scaffold for structure-specific DNA repair nucleases, also binds to the TRFH domain of TERF2.[4] The TRFH domain is responsible for other binding events, including RTEL1, and proteins that contain a TBD site.
1.2. Myb Domain
The Myb domain acts by binding to double-stranded telomeric DNA. This region gets its name from a viral protein called Myb derived from the avian myeloblastosis virus. Specifically, the sequence that this Myb domain targets on the DNA is (GGTTAG/CCAATC)n.
1.3. Basic and Hinge Domains
Two other domains also work to bind and influence the activity of proteins associated with the TERF2 protein. The basic domain sits at the N-terminal, and has two main functions: the prevention of t-loop excision by XRCC3, and the inhibition of SLX4. The final domain of TERF2 is called the hinge domain. This domain contains a motif for binding the shelterin protein TIN2, which acts as a stabilizing protein, connecting units that are attached to double stranded and single stranded DNA.[5] This domain also is responsible for binding to RAP1, and helps to inhibit RNF168 recruitment at telomeres.
2. Function
This protein is present at telomeres in metaphase of the cell cycle, is a second negative regulator of telomere length, and plays a key role in the protective activity of telomeres. While having similar telomere binding activity and domain organization, TERF2 differs from TERF1 in that its N terminus is basic rather than acidic.[2]
2.1. T-Loop Formation

Telomeric ends are structurally similar to double-stranded breaks on the chromosome. To prevent the cellular DNA repair machinery from mistakenly identifying telomeres as chromosome breaks, t-loops are formed in which the 3’ TTAGGG overhang of the telomere loops back into the DNA duplex. TERF2 promotes t-loop formation by preferentially binding to a telomeric double-stranded DNA duplex containing a 3’ TTAGGG single-stranded overhang. If the 3’ TTAGGG overhang is not present, TERF2 will not bind. Once bound, it migrates to the t-loop junction where the single-stranded overhang invades the double-stranded region upstream. No other shelterin protein has been shown to promote this process and studies have demonstrated that deletion of TERF2 prevents t-loop formation, leading to excessive loss of telomeric DNA and early cell death.[7]
2.2. ATM Kinase Prevention
TERF2 plays a central role in preventing ATM kinase DNA damage response. It binds telomeric dsDNA and prevents telomeres from activating ATM kinase. This interaction of TERF2 with ATM is believed to be relevant to the mechanism by which TERF2 blocks ATM signaling. Because of its oligomeric nature, TERF2 could potentially cross-link ATM monomers and hold the kinase in its inactive dimeric state, thereby blocking amplification of the ATM signal at an early step in its activation. However, because mutations in the TERF2 dimerization domain destabilize the protein, it has not been possible to test the contribution of TERF2 oligomerization on ATM repression directly. Removal of TERF2 induces ATM-dependent apoptosis by localizing the active, phosphorylated form of ATM to unprotected chromosome ends. Since TERF2 specifically binds at telomeres and remains there when DNA damage is induced, it is unlikely to interfere with activation of the ATM kinase at different sites of DNA damage. Therefore, TERF2 could act as a telomere-specific inhibitor of ATM kinase.[8]
2.3. TERF2 Knockout Effects
Conditional deletion of TERF2 in mice cells effectively removes the shelterin nucleoprotein complex. As a result of removing this complex, several unwanted DNA damage response pathways are activated, including ATM kinase signaling, ATR kinase signaling, non-homologous end-joining (NHEJ), alt-NHEJ, C-NHEJ, 5' resection, and homology directed repair (HDR).[9] These repair pathways (in the presence of P53 knockout and Cre) often contribute to the phenotype where chromosome ends are connected to each other in a very long chain, which can be visualized by a combination of a DAPI stain and fluorescence in situ hybridization (FISH) technique.
3. Interactions
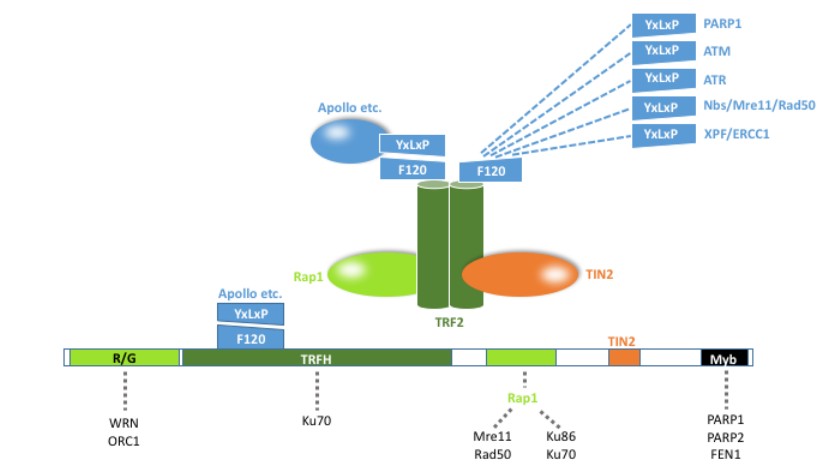
TERF2 is also known to recruit certain client proteins, also known as accessory factors. These client proteins are often recruited to TERF2 for a specific function at a specific time, often temporarily. The TRFH domain contains a F120 residue, which is the binding site of TERF2 where it recruits client proteins. These client proteins also contain a TRFH binding motif, which consists of a conserved 6-amino acid sequence of the following formula: YxLxP, where "x" can be any amino acid substituted.[10] The above-mentioned Apollo nuclease (one of many TERF2's client proteins) also contains the formulaic motif; its specific motif sequence is YLLTP.
TERF1 also demonstrates similar client protein recruitment mechanism as TERF2, except that it diverges at two concepts: 1) the TRFH of TERF1 contains a F142 residue, 2) the client proteins specific for TERF1 contain the TRFH binding motif sequence of FxLxP, where the amino acid Y (tyrosine) is replaced with F (phenylalanine).
TERF2 has also been shown to interact with:
- Ku70,[11]
- MRE11A,[12]
- Nibrin[12] and
- Rad50,[12][13]
- TERF2IP,[12][13][14] and
- Werner syndrome ATP-dependent helicase.[15]
4. Disease Relevance of TERF2
4.1. Cancer
Telomerase is an enzyme that works to create telomeric ends for DNA, and it is thought to play important roles in the development of cancer. Specifically, telomeric stability is known to be a common occurrence in cancer cells.[16] Along with the telomerase, the shelterin complex, and TERF2 and TERF1 specifically, also have been noted to control the lengths of telomeres formed by these telomerases. Shelterin works to protect telomeres against unsuitable activation of the DNA damage response pathway, as noted in the function section above. TERF2 as part of the shelterin complex, has been known to block the ATM signaling pathways and prevent chromosome end fusion. In cancer cells, TERF2 phosphorylation by extracellular signal-regulated kinase (ERK1/2) is a controlling factor in the major pro-oncogenic signaling pathways (RAS/RAF/MEK/ERK) that affect telomeric stability.[16] Additionally, when TERF2 was non-phosphorylated in melanoma cells, there was a cell induced DNA damage response, arresting growth and causing tumor reversion.[16] Studies have found that in tumor cells, TERF2 levels are observed to be high, and this raised level of TERF2 contributes to oncogenesis in a variety of ways.[17][18][19] This high level of TERF2 decreases the ability to recruit and activate natural killer cells in human tumor cells.[17] One study used a dominant negative form of TERF2ΔBΔC, to inhibit TERF2, and found that it could induce a reversion malignant phenotype in human melanoma cells.[18] Therefore, over-expression of TERF2ΔBΔC, and therefore blocking of TERF2, induced apoptosis and reduced tumourigenicity in certain cell lines.[18] Additionally, upregulation of TERF2 may be the cause of the establishment and maintenance of short telomeres.[19] These short telomeres increase chromosomal instability, and increase the chances of certain cancers progressing in the body, such as with leukemia.[19] In gastric mucosa tissues, the expression of TERF2 proteins was significantly higher than normal, and this over-expression of TERF2, along with over-expression of TERF1, TIN2, TERT, and BRCA1 protein transposition, may cause a reduction in telomere length, further contributing to multistage carcinogenesis of gastric cancer.[20]
The content is sourced from: https://handwiki.org/wiki/Biology:TERF2
References
- "Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2". Nat. Genet. 17 (2): 231–5. November 1997. doi:10.1038/ng1097-231. PMID 9326950. https://dx.doi.org/10.1038%2Fng1097-231
- "Entrez Gene: TERF2 telomeric repeat binding factor 2". https://www.ncbi.nlm.nih.gov/sites/entrez?Db=gene&Cmd=ShowDetailView&TermToSearch=7014.
- "Apollo Contributes to G Overhang Maintenance and Protects Leading-End Telomeres". Molecular Cell 39 (4): 606–617. 27 August 2010. doi:10.1016/j.molcel.2010.06.031. http://www.sciencedirect.com/science/article/pii/S1097276510004983.
- "Localization-Dependent and -Independent Roles of SLX4 in Regulating Telomeres". Cell Press 4 (5): 853–860. 12 September 2013. doi:10.1016/j.celrep.2013.07.033. http://www.sciencedirect.com/science/article/pii/S2211124713003951.
- Palm, Wilhelm; de Lange, Titia (2008-11-04). "How Shelterin Protects Mammalian Telomeres". Annual Review of Genetics 42 (1): 301–334. doi:10.1146/annurev.genet.41.110306.130350. ISSN 0066-4197. http://www.annualreviews.org/doi/10.1146/annurev.genet.41.110306.130350.
- Arnoult, Nausica; Karlseder, Jan (2015-11-01). "Complex interactions between the DNA-damage response and mammalian telomeres" (in en). Nature Structural & Molecular Biology 22 (11): 859–866. doi:10.1038/nsmb.3092. PMC 4739752. http://www.nature.com/nsmb/journal/v22/n11/fig_tab/nsmb.3092_F2.html.
- Stansel, Rachel M.; de Lange, Titia; Griffith, Jack D. (2001-10-01). "T-loop assembly in vitro involves binding of TRF2 near the 3′ telomeric overhang". The EMBO Journal 20 (19): 5532–5540. doi:10.1093/emboj/20.19.5532. ISSN 0261-4189. PMID 11574485. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=125642
- Karlseder, Jan; Hoke, Kristina; Mirzoeva, Olga K.; Bakkenist, Christopher; Kastan, Michael B.; Petrini, John H. J.; de Lange, Titia (2004-08-01). "The telomeric protein TRF2 binds the ATM kinase and can inhibit the ATM-dependent DNA damage response". PLOS Biology 2 (8): E240. doi:10.1371/journal.pbio.0020240. ISSN 1545-7885. PMID 15314656. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=509302
- Sfeir, Agnel; Lange, Titia de (2012-05-04). "Removal of Shelterin Reveals the Telomere End-Protection Problem" (in en). Science 336 (6081): 593–597. doi:10.1126/science.1218498. ISSN 0036-8075. PMID 22556254. PMC 3477646. http://science.sciencemag.org/content/336/6081/593.
- Chen, Yong; Yang, Yuting; Overbeek, Megan van; Donigian, Jill R.; Baciu, Paul; Lange, Titia de; Lei, Ming (2008-02-22). "A Shared Docking Motif in TRF1 and TRF2 Used for Differential Recruitment of Telomeric Proteins" (in en). Science 319 (5866): 1092–1096. doi:10.1126/science.1151804. ISSN 0036-8075. PMID 18202258. http://science.sciencemag.org/content/319/5866/1092.
- "Interaction of human Ku70 with TRF2". FEBS Lett. 481 (1): 81–5. September 2000. doi:10.1016/s0014-5793(00)01958-x. PMID 10984620. https://dx.doi.org/10.1016%2Fs0014-5793%2800%2901958-x
- "Cell-cycle-regulated association of RAD50/MRE11/NBS1 with TRF2 and human telomeres". Nat. Genet. 25 (3): 347–52. July 2000. doi:10.1038/77139. PMID 10888888. https://dx.doi.org/10.1038%2F77139
- "The human Rap1 protein complex and modulation of telomere length". J. Biol. Chem. 279 (27): 28585–91. July 2004. doi:10.1074/jbc.M312913200. PMID 15100233. https://dx.doi.org/10.1074%2Fjbc.M312913200
- "Identification of human Rap1: implications for telomere evolution". Cell 101 (5): 471–83. May 2000. doi:10.1016/S0092-8674(00)80858-2. PMID 10850490. https://dx.doi.org/10.1016%2FS0092-8674%2800%2980858-2
- "Telomere-binding protein TRF2 binds to and stimulates the Werner and Bloom syndrome helicases". J. Biol. Chem. 277 (43): 41110–9. October 2002. doi:10.1074/jbc.M205396200. PMID 12181313. https://dx.doi.org/10.1074%2Fjbc.M205396200
- Picco, Vincent; Coste, Isabelle; Giraud-Panis, Marie-Josèphe; Renno, Toufic; Gilson, Eric; Pagès, Gilles (2016-06-29). "ERK1/2/MAPK pathway-dependent regulation of the telomeric factor TRF2". Oncotarget. doi:10.18632/oncotarget.10316. ISSN 1949-2553. PMID 27366950. https://dx.doi.org/10.18632%2Foncotarget.10316
- Biroccio, Annamaria; Cherfils-Vicini, Julien; Augereau, Adeline; Pinte, Sébastien; Bauwens, Serge; Ye, Jing; Simonet, Thomas; Horard, Béatrice et al. (2013-07-01). "TRF2 inhibits a cell-extrinsic pathway through which natural killer cells eliminate cancer cells" (in en). Nature Cell Biology 15 (7): 818–828. doi:10.1038/ncb2774. ISSN 1465-7392. PMID 23792691. http://www.nature.com/ncb/journal/v15/n7/abs/ncb2774.html.
- Biroccio, Annamaria (August 2006). "TRF2 inhibition triggers apoptosis and reduces tumourigenicity of human melanoma cells". European Journal of Cancer 42: 1881–1888. doi:10.1016/j.ejca.2006.03.010. http://www.sciencedirect.com/science/article/pii/S0959804906003339.
- Bellon, Marcia; Datta, Abhik; Brown, Megan; Pouliquen, Jean-Francois; Couppie, Pierre; Kazanji, Mirdad; Nicot, Christophe (2006-11-01). "Increased expression of telomere length regulating factors TRF1, TRF2 and TIN2 in patients with adult T-cell leukemia" (in en). International Journal of Cancer 119 (9): 2090–2097. doi:10.1002/ijc.22026. ISSN 1097-0215. PMID 16786598. http://onlinelibrary.wiley.com/doi/10.1002/ijc.22026/abstract.
- Hu, Hua; Zhang, Yang; Zou, Mei; Yang, Shuai; Liang, Xiao-Qiu (2010-02-02). "Expression of TRF1, TRF2, TIN2, TERT, KU70, and BRCA1 proteins is associated with telomere shortening and may contribute to multistage carcinogenesis of gastric cancer" (in en). Journal of Cancer Research and Clinical Oncology 136 (9): 1407–1414. doi:10.1007/s00432-010-0795-x. ISSN 0171-5216. https://link.springer.com/article/10.1007/s00432-010-0795-x.