thumb|A Hanford scientist uses an Auger electron spectrometer to determine the elemental composition of surfaces. Auger electron spectroscopy (AES; pronounced [oʒe] in French) is a common analytical technique used specifically in the study of surfaces and, more generally, in the area of materials science. Underlying the spectroscopic technique is the Auger effect, as it has come to be called, which is based on the analysis of energetic electrons emitted from an excited atom after a series of internal relaxation events. The Auger effect was discovered independently by both Lise Meitner and Pierre Auger in the 1920s. Though the discovery was made by Meitner and initially reported in the journal Zeitschrift für Physik in 1922, Auger is credited with the discovery in most of the scientific community. Until the early 1950s Auger transitions were considered nuisance effects by spectroscopists, not containing much relevant material information, but studied so as to explain anomalies in X-ray spectroscopy data. Since 1953 however, AES has become a practical and straightforward characterization technique for probing chemical and compositional surface environments and has found applications in metallurgy, gas-phase chemistry, and throughout the microelectronics industry.
- metallurgy
- microelectronics
- elemental composition
1. Electron Transitions and the Auger Effect
The Auger effect is an electronic process at the heart of AES resulting from the inter- and intrastate transitions of electrons in an excited atom. When an atom is probed by an external mechanism, such as a photon or a beam of electrons with energies in the range of several eV to 50 keV, a core state electron can be removed leaving behind a hole. As this is an unstable state, the core hole can be filled by an outer shell electron, whereby the electron moving to the lower energy level loses an amount of energy equal to the difference in orbital energies. The transition energy can be coupled to a second outer shell electron, which will be emitted from the atom if the transferred energy is greater than the orbital binding energy.[1][2][3][4][5][6] An emitted electron will have a kinetic energy of:
- [math]\displaystyle{ E_{\text{kin}}=E_{\text{Core State}}-E_B-E_{C}' }[/math]
where [math]\displaystyle{ E_{\text{Core State}} }[/math], [math]\displaystyle{ E_B }[/math], [math]\displaystyle{ E_C' }[/math] are respectively the core level, first outer shell, and second outer shell electron binding energies (measured from the vacuum level) which are taken to be positive. The apostrophe (tic) denotes a slight modification to the binding energy of the outer shell electrons due to the ionized nature of the atom; often however, this energy modification is ignored in order to ease calculations.[2][7] Since orbital energies are unique to an atom of a specific element, analysis of the ejected electrons can yield information about the chemical composition of a surface. Figure 1 illustrates two schematic views of the Auger process.
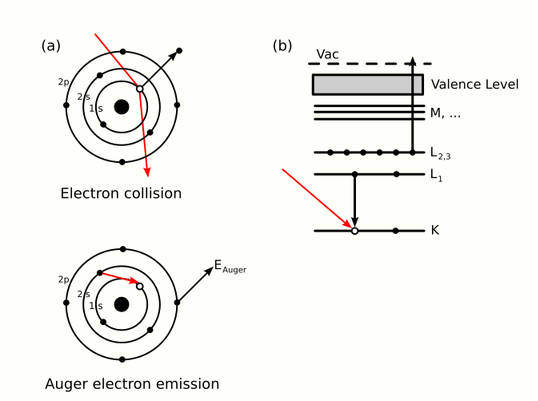
The types of state-to-state transitions available to electrons during an Auger event are dependent on several factors, ranging from initial excitation energy to relative interaction rates, yet are often dominated by a few characteristic transitions. Because of the interaction between an electron's spin and orbital angular momentum (spin-orbit coupling) and the concomitant energy level splitting for various shells in an atom, there are a variety of transition pathways for filling a core hole. Energy levels are labeled using a number of different schemes such as the j-j coupling method for heavy elements (Z ≥ 75), the Russell-Saunders L-S method for lighter elements (Z < 20), and a combination of both for intermediate elements.[2][8][9] The j-j coupling method, which is historically linked to X-ray notation, is almost always used to denote Auger transitions. Thus for a [math]\displaystyle{ KL_1L_{2,3} }[/math] transition, [math]\displaystyle{ K }[/math] represents the core level hole, [math]\displaystyle{ L_1 }[/math] the relaxing electron's initial state, and [math]\displaystyle{ L_{2,3} }[/math] the emitted electron's initial energy state. Figure 1(b) illustrates this transition with the corresponding spectroscopic notation. The energy level of the core hole will often determine which transition types will be favored. For single energy levels, i.e. K, transitions can occur from the L levels, giving rise to strong KLL type peaks in an Auger spectrum. Higher level transitions can also occur, but are less probable. For multi-level shells, transitions are available from higher energy orbitals (different n, ℓ quantum numbers) or energy levels within the same shell (same n, different ℓ number).[1] The result are transitions of the type LMM and KLL along with faster Coster–Kronig transitions such as LLM.[1][2] While Coster–Kronig transitions are faster, they are also less energetic and thus harder to locate on an Auger spectrum. As the atomic number Z increases, so too does the number of potential Auger transitions. Fortunately, the strongest electron-electron interactions are between levels that are close together, giving rise to characteristic peaks in an Auger spectrum. KLL and LMM peaks are some of the most commonly identified transitions during surface analysis.[2] Finally, valence band electrons can also fill core holes or be emitted during KVV-type transitions.
Several models, both phenomenological and analytical, have been developed to describe the energetics of Auger transitions. One of the most tractable descriptions, put forth by Jenkins and Chung, estimates the energy of Auger transition ABC as:
- [math]\displaystyle{ E_{ABC}=E_A(Z)-0.5[E_B(Z)+E_B(Z+1)]-0.5[E_C(Z)+E_C(Z+1)] }[/math]
[math]\displaystyle{ E_i(Z) }[/math] are the binding energies of the [math]\displaystyle{ i }[/math]th level in element of atomic number Z and [math]\displaystyle{ E_i(Z+1) }[/math] are the energies of the same levels in the next element up in the periodic table. While useful in practice, a more rigorous model accounting for effects such as screening and relaxation probabilities between energy levels gives the Auger energy as:
- [math]\displaystyle{ E_{ABC}=E_A-E_B-E_C-F(BC:x)+R_{xin}+R_{xex} }[/math]
where [math]\displaystyle{ F(BC:x) }[/math] is the energy of interaction between the B and C level holes in a final atomic state x and the R's represent intra- and extra-atomic transition energies accounting for electronic screening.[2] Auger electron energies can be calculated based on measured values of the various [math]\displaystyle{ E_i }[/math] and compared to peaks in the secondary electron spectrum in order to identify chemical species. This technique has been used to compile several reference databases used for analysis in current AES setups.
2. Experimental Setup and Quantification
2.1. Instrumentation

Surface sensitivity in AES arises from the fact that emitted electrons usually have energies ranging from 50 eV to 3 keV and at these values, electrons have a short mean free path in a solid. The escape depth of electrons is therefore localized to within a few nanometers of the target surface, giving AES an extreme sensitivity to surface species.[6] Because of the low energy of Auger electrons, most AES setups are run under ultra-high vacuum (UHV) conditions. Such measures prevent electron scattering off of residual gas atoms as well as the formation of a thin "gas (adsorbate) layer" on the surface of the specimen, which degrades analytical performance.[5][6] A typical AES setup is shown schematically in figure 2. In this configuration, focused electrons are incident on a sample and emitted electrons are deflected into a cylindrical mirror analyzer (CMA). In the detection unit, Auger electrons are multiplied and the signal sent to data processing electronics. Collected Auger electrons are plotted as a function of energy against the broad secondary electron background spectrum. The detection unit and data processing electronics are collectively referred to as the electron energy analyzer[10].
Since the intensity of the Auger peaks may be small compared to the noise level of the background, AES is often run in a derivative mode that serves to highlight the peaks by modulating the electron collection current via a small applied AC voltage. Since this [math]\displaystyle{ \Delta V=k\sin(\omega t) }[/math], the collection current becomes [math]\displaystyle{ I(V+k\sin(\omega t)) }[/math]. Taylor expanding gives:
- [math]\displaystyle{ I(V+k\sin(\omega t))\approx I_0+I'(V+k\sin(\omega t))+O(I'') }[/math]
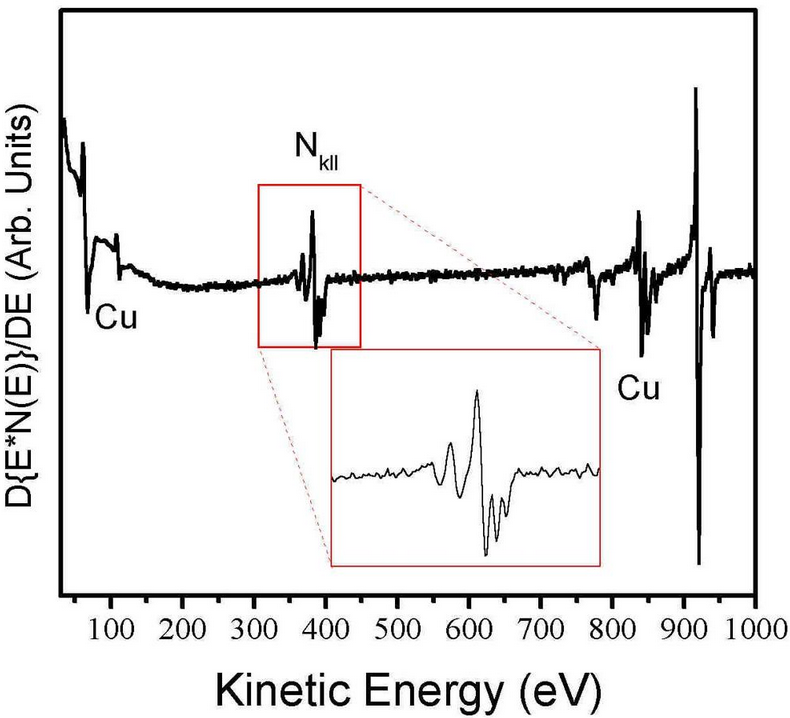
Using the setup in figure 2, detecting the signal at frequency ω will give a value for [math]\displaystyle{ I' }[/math] or [math]\displaystyle{ \frac{dN}{dE} }[/math].[5][6] Plotting in derivative mode also emphasizes Auger fine structure, which appear as small secondary peaks surrounding the primary Auger peak. These secondary peaks, not to be confused with high energy satellites, which are discussed later, arise from the presence of the same element in multiple different chemical states on a surface (i.e. Adsorbate layers) or from relaxation transitions involving valence band electrons of the substrate. Figure 3 illustrates a derivative spectrum from a copper nitride film clearly showing the Auger peaks. The peak in derivative mode is not the true Auger peak, but rather the point of maximum slope of N(E), but this concern is usually ignored.[6]
2.2. Quantitative Analysis
Semi-quantitative compositional and element analysis of a sample using AES is dependent on measuring the yield of Auger electrons during a probing event. Electron yield, in turn, depends on several critical parameters such as electron-impact cross-section and fluorescence yield.[3][5] Since the Auger effect is not the only mechanism available for atomic relaxation, there is a competition between radiative and non-radiative decay processes to be the primary de-excitation pathway. The total transition rate, ω, is a sum of the non-radiative (Auger) and radiative (photon emission) processes. The Auger yield, [math]\displaystyle{ \omega_A }[/math], is thus related to the fluorescence (x-ray) yield, [math]\displaystyle{ \omega_X }[/math], by the relation,
- [math]\displaystyle{ \omega_A=1-\omega_X=1-\frac{W_X}{W_X+W_A} }[/math]
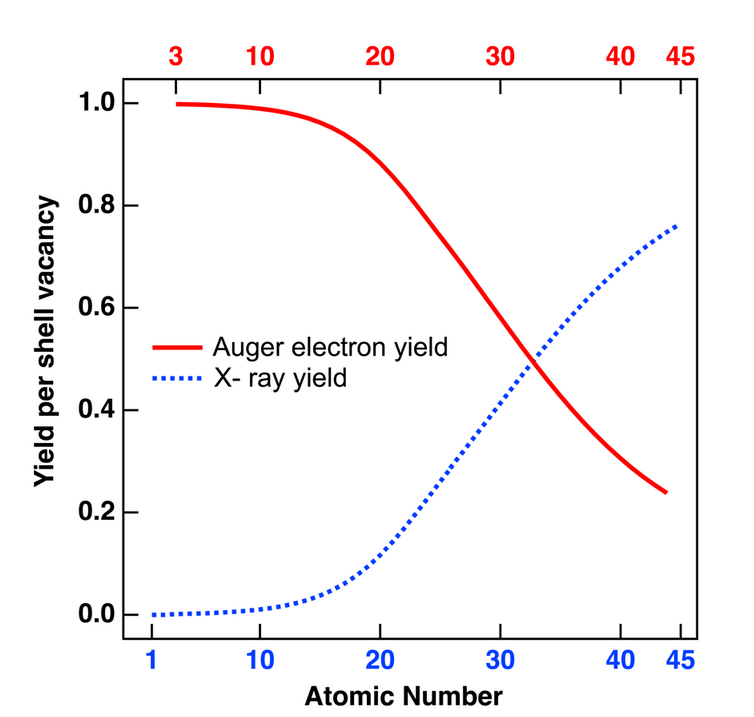
where [math]\displaystyle{ W_X }[/math] is the X-ray transition probability and [math]\displaystyle{ W_A }[/math] is the Auger transition probability.[5] Attempts to relate the fluorescence and Auger yields to atomic number have resulted in plots similar to figure 4. A clear transition from electron to photon emission is evident in this chart for increasing atomic number. For heavier elements, x-ray yield becomes greater than Auger yield, indicating an increased difficulty in measuring the Auger peaks for large Z-values. Conversely, AES is sensitive to the lighter elements, and unlike X-ray fluorescence, Auger peaks can be detected for elements as light as lithium (Z = 3). Lithium represents the lower limit for AES sensitivity since the Auger effect is a "three state" event necessitating at least three electrons. Neither H nor He can be detected with this technique. For K-level based transitions, Auger effects are dominant for Z < 15 while for L- and M-level transitions, AES data can be measured for Z ≤ 50.[5] The yield limits effectively prescribe a cutoff for AES sensitivity, but complex techniques can be utilized to identify heavier elements, such as uranium and americium, using the Auger effect.[11]
Another critical quantity that determines yield of Auger electrons at a detector is the electron impact cross-section. Early approximations (in cm2) of the cross-section were based on the work of Worthington and Tomlin,
- [math]\displaystyle{ \sigma_{ax}(E)=1.3\times10^{13}b\frac{C}{E_p} }[/math]
with b acting as a scaling factor between 0.25 and 0.35, and C a function of the primary electron beam energy, [math]\displaystyle{ E_p }[/math]. While this value of [math]\displaystyle{ \sigma_{ax} }[/math] is calculated for an isolated atom, a simple modification can be made to account for matrix effects:
- [math]\displaystyle{ \sigma (E)=\sigma_{ax}[1+r_m(E_p,\alpha)] }[/math]
where α is the angle to the surface normal of the incident electron beam; rm can be established empirically and encompasses electron interactions with the matrix such as ionization due to backscattered electrons. Thus the total yield can be written as:
- [math]\displaystyle{ Y(t)=N_x \times \delta t \times \sigma (E,t )[1-\omega_X] \exp\left(-t\cos \frac{\theta}{\lambda}\right) \times I(t)\times T\times\frac{d(\Omega)}{4\pi} }[/math]
Here Nx is the number of x atoms per volume, λ the electron escape depth, θ the analyzer angle, T the transmission of the analyzer, I(t) the electron excitation flux at depth t, dΩ the solid angle, and δt is the thickness of the layer being probed. Encompassed in these terms, especially the Auger yield, which is related to the transition probability, is the quantum mechanical overlap of the initial and final state wave functions. Precise expressions for the transition probability, based on first-order perturbation Hamiltonians, can be found in Thompson and Baker.[3] Often, all of these terms are not known, so most analyses compare measured yields with external standards of known composition. Ratios of the acquired data to standards can eliminate common terms, especially experimental setup characteristics and material parameters, and can be used to determine element composition.[2][5][6] Comparison techniques work best for samples of homogeneous binary materials or uniform surface layers, while elemental identification is best obtained from comparison of pure samples.
3. Uses
There are a number of electron microscopes that have been specifically designed for use in Auger spectroscopy; these are termed scanning Auger microscopes (SAMs) and can produce high resolution, spatially resolved chemical images.[2][4][6][11][12] SAM images are obtained by stepping a focused electron beam across a sample surface and measuring the intensity of the Auger peak above the background of scattered electrons. The intensity map is correlated to a gray scale on a monitor with whiter areas corresponding to higher element concentration. In addition, sputtering is sometimes used with Auger spectroscopy to perform depth profiling experiments. Sputtering removes thin outer layers of a surface so that AES can be used to determine the underlying composition.[2][3][4][5] Depth profiles are shown as either Auger peak height vs. sputter time or atomic concentration vs. depth. Precise depth milling through sputtering has made profiling an invaluable technique for chemical analysis of nanostructured materials and thin films. AES is also used extensively as an evaluation tool on and off fab lines in the microelectronics industry, while the versatility and sensitivity of the Auger process makes it a standard analytical tool in research labs.[13][14][15][16] Theoretically, Auger spectra can also be utilized to distinguish between protonation states. When a molecule is protonated or deprotonated, the geometry and electronic structure is changed, and AES spectra reflect this. In general, as a molecule becomes more protonated, the ionization potentials increase and the kinetic energy of the emitted outer shell electrons decreases.[17]
Despite the advantages of high spatial resolution and precise chemical sensitivity attributed to AES, there are several factors that can limit the applicability of this technique, especially when evaluating solid specimens. One of the most common limitations encountered with Auger spectroscopy are charging effects in non-conducting samples.[1][2] Charging results when the number of secondary electrons leaving the sample is different from the number of incident electrons, giving rise to a net positive or negative electric charge at the surface. Both positive and negative surface charges severely alter the yield of electrons emitted from the sample and hence distort the measured Auger peaks. To complicate matters, neutralization methods employed in other surface analysis techniques, such as secondary ion mass spectrometry (SIMS), are not applicable to AES, as these methods usually involve surface bombardment with either electrons or ions (i.e. flood gun). Several processes have been developed to combat the issue of charging, though none of them is ideal and still make quantification of AES data difficult.[2][5] One such technique involves depositing conductive pads near the analysis area to minimize regional charging. However, this type of approach limits SAM applications as well as the amount of sample material available for probing. A related technique involves thinning or "dimpling" a non-conductive layer with Ar+ ions and then mounting the sample to a conductive backing prior to AES.[18][19] This method has been debated, with claims that the thinning process leaves elemental artifacts on a surface and/or creates damaged layers that distort bonding and promote chemical mixing in the sample. As a result, the compositional AES data is considered suspect. The most common setup to minimize charging effects includes use of a glancing angle (~10°) electron beam and a carefully tuned bombarding energy (between 1.5 keV and 3 keV). Control of both the angle and energy can subtly alter the number of emitted electrons vis-à-vis the incident electrons and thereby reduce or altogether eliminate sample charging.[1][4][5]
In addition to charging effects, AES data can be obscured by the presence of characteristic energy losses in a sample and higher order atomic ionization events. Electrons ejected from a solid will generally undergo multiple scattering events and lose energy in the form of collective electron density oscillations called plasmons.[1][6] If plasmon losses have energies near that of an Auger peak, the less intense Auger process may become dwarfed by the plasmon peak. As Auger spectra are normally weak and spread over many eV of energy, they are difficult to extract from the background and in the presence of plasmon losses; deconvolution of the two peaks becomes extremely difficult. For such spectra, additional analysis through chemical sensitive surface techniques like x-ray photoelectron spectroscopy (XPS) is often required to disentangle the peaks.[1] Sometimes an Auger spectrum can also exhibit "satellite" peaks at well-defined off-set energies from the parent peak. Origin of the satellites is usually attributed to multiple ionization events in an atom or ionization cascades in which a series of electrons is emitted as relaxation occurs for core holes of multiple levels.[1][2] The presence of satellites can distort the true Auger peak and/or small peak shift information due to chemical bonding at the surface. Several studies have been undertaken to further quantify satellite peaks.[20]
Despite these sometimes substantial drawbacks, Auger electron spectroscopy is a widely used surface analysis technique that has been successfully applied to many diverse fields ranging from gas phase chemistry to nanostructure characterization. Very new class of high-resolving electrostatic energy analyzers recently developed – the face-field analyzers (FFA)[21] can be used for remote electron spectroscopy of distant surfaces or surfaces with large roughness or even with deep dimples. These instruments are designed as if to be specifically used in combined scanning electron microscopes (SEMs). "FFA" in principle have no perceptible end-fields, which usually distort focusing in most of analysers known, for example, well known CMA.
Sensitivity, quantitative detail, and ease of use have brought AES from an obscure nuisance effect to a functional and practical characterization technique in just over fifty years. With applications both in the research laboratory and industrial settings, AES will continue to be a cornerstone of surface-sensitive electron-based spectroscopies.
The content is sourced from: https://handwiki.org/wiki/Auger_electron_spectroscopy
References
- Thomas A., Carlson (1975). Photoelectron and Auger Spectroscopy. New York: Plenum Press. ISBN 0-306-33901-3.
- Briggs, David; Martin P. Seah (1983). Practical Surface Analysis by Auger and X-ray Photoelectron Spectroscopy. Chichester: John Wiley & Sons. ISBN 0-471-26279-X. https://www.wikipedia.org/wiki/John%20Wiley%20%26%20Sons
- Thompson, Michael; M. D. Baker; A. Christie; J. F. Tyson (1985). Auger Electron Spectroscopy. Chichester: John Wiley & Sons. ISBN 0-471-04377-X.
- Davis, L. E. (ed.) (1980). Modern Surface Analysis: Metallurgical Applications of Auger Electron Spectroscopy (AES) and X-ray Photoelectron Spectroscopy (XPS). Warrendale: The Metallurgical Society of AIME. ISBN 0-89520-358-8.
- Feldman, Leonard C.; James W. Mayer (1986). Fundamentals of Surface and Thin Film Analysis. Upper Saddle River: Prentice Hall. ISBN 0-13-500570-1.
- Oura, K.; V. G. Lifshits; A. A. Saranin; A. V. Zotov; M. Katayama (2003). Surface Science: An Introduction. Berlin: Springer. ISBN 3-540-00545-5.
- Auger spectroscopy National Physical Laboratory: Kaye & Laby, Tables of Physical and Chemical Constants http://www.kayelaby.npl.co.uk/atomic_and_nuclear_physics/4_1/4_1_3.html
- Kittel, Charles (1996). Introduction to Solid State Physics (7th ed.). New York: John Wiley & Sons. ISBN 81-265-1045-5.
- Ashcroft, Neil; Mermin, N. David (1976). Solid State Physics. Ithaca: Thomson Learning. ISBN 0-03-049346-3.
- "Auger Electron Spectroscopy". Physical Electronics, Inc..(PHI). 2020. https://www.phi.com/surface-analysis-techniques/aes.html.
- Grant, John T.; David Briggs (2003). Surface Analysis by Auger and X-ray Photoelectron Spectroscopy. Chichester: IM Publications. ISBN 1-901019-04-7.
- Attard, Gary; Barnes, Colin. Surfaces. Oxford Chemistry Primers. p. 47. ISBN 978-0-19-855686-2.
- Chao, Liang-Chiun; Shih-Hsuan Yang (June 2007). "Growth and Auger electron spectroscopy characterization of donut-shaped ZnO nanostructures". Applied Surface Science 253 (17): 7162–7165. doi:10.1016/j.apsusc.2007.02.184. Bibcode: 2007ApSS..253.7162C. https://dx.doi.org/10.1016%2Fj.apsusc.2007.02.184
- Soohwan Jang et al. (May 2007). "Comparison of E-beam and Sputter-Deposited ITO Films for 1.55 μm Metal–Semiconductor–Metal Photodetector Applications". Journal of the Electrochemical Society 154 (5): H336–H339. doi:10.1149/1.2667428. https://dx.doi.org/10.1149%2F1.2667428
- Mingjie Xu et al. (March 2006). "Biomimetic silicification of 3D polyamine-rich scaffolds assembled by direct ink writing". Soft Matter 2 (3): 205–209. doi:10.1039/b517278k. Bibcode: 2006SMat....2..205X. https://dx.doi.org/10.1039%2Fb517278k
- Gondran, Carolyn F. H.; Charlene Johnson; Kisik Choi (September 2006). "Front and back side Auger electron spectroscopy depth profile analysis to verify an interfacial reaction at the HfN/SiO2 interface". Journal of Vacuum Science and Technology B 24 (5): 2457. doi:10.1116/1.2232380. Bibcode: 2006JVSTB..24.2457G. https://dx.doi.org/10.1116%2F1.2232380
- Kryzhevoi, Nikolai V., and Lorenz S. Cederbaum. "Exploring Protonation and Deprotonation Effects with Auger Electron Spectroscopy." The Journal of Physical Chemistry Letters. 3.18 (2012): 2733-737.
- Yu, Ling; Deling Jin (April 2001). "AES and SAM microanalysis of structure ceramics by thinning and coating the backside". Surface and Interface Analysis 31 (4): 338–342. doi:10.1002/sia.982. https://dx.doi.org/10.1002%2Fsia.982
- Cazaux, Jacques (December 1992). "Mechanisms of charging in electron spectroscopy". Journal of Electronic Spectroscopy and Related Phenomena 105 (2–3): 155–185. doi:10.1016/S0368-2048(99)00068-7. https://dx.doi.org/10.1016%2FS0368-2048%2899%2900068-7
- Went, M. R.; M. Vos; A. S. Kheifets (November 2006). "Satellite structure in Auger and (e,2e) spectra of germanium". Radiation Physics and Chemistry 75 (11): 1698–1703. doi:10.1016/j.radphyschem.2006.09.003. Bibcode: 2006RaPC...75.1698W. https://dx.doi.org/10.1016%2Fj.radphyschem.2006.09.003
- Ilyin, A (2003). "New class of electrostatic energy analyzers with a cylindrical face-field". Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment 500: 62. doi:10.1016/S0168-9002(03)00334-6. Bibcode: 2003NIMPA.500...62I. https://dx.doi.org/10.1016%2FS0168-9002%2803%2900334-6