1. Involvement of Wnt Signaling in Chronic Pain
In the last decade, a growing body of evidence has demonstrated the involvement of the Wnt signaling pathway in the context of chronic pain, both in patients and in several pre-clinical mouse models of pain [
26].
Wnt signaling is activated at different levels along the pain pathway and in diverse cell types, depending on the chronic pain condition or the pain model studied (Figure 1). Most studies, using preclinical pain models, focused on Wnt pathway activation in dorsal root ganglia (DRGs) and the spinal cord. In contrast, reports considering the supraspinal level are almost completely absent.
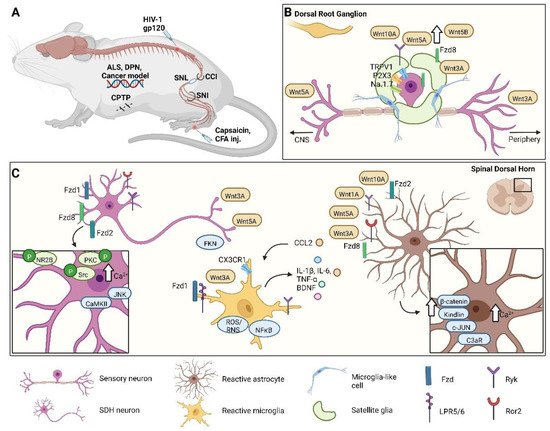
Figure 1. Wnt’s contributions to different pain conditions in various neuronal and non-neuronal cells. (A) Schematic representation of different pain models in mice. (B) After neuropathic pain induction, in dorsal root ganglia, activated nociceptors increase the receptors’ expression, such as TRPV1, P2X3, Na1.7, Ryk, and Fzd8, and that of Wnt-related proteins, e.g., Wnt5a, Wnt5b, Wnt3A, and Wnt10a. Likewise, in satellite cells, Fdz8 and Wnt3a are also overexpressed. (C) In the spinal dorsal horn, Wnt signaling is involved in pain sensation and acts on neuronal and non-neuronal cells. Many Fzd receptors and co-receptors (Ryk and Ror2) are upregulated in neurons, which brings about phosphorylation, activation of downstream targets (NR2B, Src, and PKC), and a Ca2+ increase. This results in increases in JNK and CaMKII and the release of Wnt3a, Wnt5a, and FKN. Astrocytes harbor large amounts of Wnt proteins and receptors, which are upregulated during pain. As a consequence, the concentration of Ca2+ rises in the cytoplasm, along with the concentrations of β-catenin, kindlin, c-JUN, and C3aR; and the release of CCL2 increases. This last chemokine triggers microglia, which in situations of pain upregulates Fzd, the LRP5/6 co-receptor, and Wnt3a. Reactive microglia increase the expression of CX3CR1, ROS/RNS, and NF-kB and the secretion of IL-1β, IL-6, TNF-α, and BDNF, which in turn escalate the inflammatory condition. ALS, amyotrophic lateral sclerosis; DPN, diabetic peripheral neuropathy; CPTP, chronic post-thoracotomy pain; CCI, chronic constriction injury; SNL, spinal nerve ligation; SNI, spared nerve injury; CFA, complete Freund’s adjuvant; inj., injection; Fzd, Frizzled; FKN, Fractalkine; P, phosphorylation; NR2B, N-methyl-D-aspartate receptor subunit 2B; JNK, c-Jun amino (N)-terminal kinase; Src, Proto-oncogene tyrosine-protein kinase; PKC, protein kinase C; CaMKII, Ca2+/calmodulin-dependent protein kinase II; CCL2/MCP1, CC-chemokine ligand 2; ROS/RNS, reactive oxygen species/reactive nitrogen species; NF-kB, Nuclear factor kappa-light-chain-enhancer of activated B cells; BDNF, brain-derived neurotrophic factor; c-JUN, transcription factor Jun; C3aR, complement component 3 fragment a receptor; LRP5/6, low-density lipoprotein receptor-related protein 5/6; Ryk, receptor-like tyrosine kinase; Ror2, receptor tyrosine kinase-like orphan receptor 2; CX3CR1, CX3C chemokine receptor 1; IL, interleukin; TNF-α, tumor necrosis factor alpha.
2. Neuronal Wnt Signaling Dysregulation in Chronic Pain
Peripheral sensitization is a mechanism underlying the development of chronic pain. Generally, it originates from small molecules released by different cell types in pathological conditions that can activate and modulate the nociceptors. These small molecules can alter the properties of nociceptive ion channels such as transient receptor potential cation channel subfamily V member 1 (TRPV1), transient receptor potential ankyrin 1 (TRPA1), voltage-dependent sodium channel (Na
v) 1.7, and Na
v1.8 via post-translational modifications such as phosphorylation, leading to increased neuronal activity, reduced activation thresholds, or increased currents [
27].
More recently, He et al. have shown that Wnt5b-Ryk signaling is involved in bone cancer pain via Ca
2+/calmodulin-dependent protein kinase II (CaMKII)-dependent, P2X3-mediated increased DRG excitability [
30]. In a mouse model of diabetic neuropathic pain (DNP), Wnt5a is released from A-fiber in a Wnt ligand secretion mediator (GPR177)-dependent way and directly binds and activates TRPV1 receptors expressed by the neighboring C-fibers [
31]. Interestingly, in two different rat models of neuropathic pain, paclitaxel-induced and streptozotocin (STZ)-induced pain, Wnt signaling pathway inhibitors NSC668036 and PNU74654 reverse the reduction in intraepidermal nerve fiber density (IENFD) [
32,
33], used as a clinical marker of chemotherapy-induced peripheral neuropathy [
34] and diabetic peripheral neuropathy [
35]. These findings underline the importance of Wnt signaling in mediating neuropathic pain also at the level of peripheral terminals, although the study of this phenomenon is only beginning.
In cancer conditions and different models of chronic pain, such as tumor-cell-induced pain (TCI), chemotherapy-induced neuropathic pain (PTX-induced pain), and chronic constriction injury (CCI, a model of neuropathic pain with a strong inflammatory component), Wnt ligands such as Wnt3a, Wnt5b, and Wnt10a; Wnt receptors such as Fzd8 and Ryk; and signaling molecules such as β-catenin, GSK-3β, and TCF4 are upregulated in DRGs [
29,
36].
The increased expression of Wnt ligands in DRG supports their release from the sensory afferents into the spinal cord in an activity-dependent manner driven by pain stimuli [
29,
37]. Indeed, neuronal activity controls both the expression and the secretion of Wnts (reviewed in [
38]).
At the spinal cord level, Wnt signaling can modulate the pain sensation by acting directly on neurons and regulating synaptic plasticity, or by recruiting non-neuronal cells such as microglia and astrocytes. Wnt ligands are known to be important modulators of synaptic plasticity [
39], a phenomenon that is well established as a crucial mechanism underlying chronic pain [
40,
41]. Activation of Wnt signaling via NMDA-receptor-mediated synaptic Wnt3a release induces LTP, a form of synaptic plasticity [
25]. Moreover, the inhibition of Wnt signaling blocks LTP. Accordingly, Wnt family members are upregulated into the spinal cord in several chronic pain conditions and mouse pain models.
Activation of the Wnt/β-catenin canonical pathway leads to the increased production and secretion of pro-inflammatory cytokines and BDNF, which enhance neuronal excitability and synaptic plasticity [
28,
41,
42]. Furthermore, the canonical pathway regulates the N-methyl-D-aspartate receptor subunit 2B (NR2B)- and Ca
2+-dependent signals in the dorsal horn [
29]. In the CCI and spinal nerve ligation (SNL) neuropathic pain models, Wnt5b/Ryk signaling contributes to the development of neuropathic pain; these proteins are upregulated in DRGs and the spinal cord after nerve injury [
42]. Ror2 plays a relevant role in CCI-induced neuropathic pain: modulating synaptic plasticity via phosphorylation of NR2B, protein kinase C (PKC), and Src family kinases in the spinal cord [
43]. Furthermore, in models of nerve injury or inflammatory pain, Wnt5a is secreted in an activity-dependent manner and mediates chronic pain via modulation of synaptic spines [
37]. Importantly, blocking Wnt pathways is sufficient to reduce neuropathic pain. Indeed, blocking Ryk signaling decreases neuronal excitability, lowers the enhanced synaptic plasticity between C-fibers and dorsal horn neurons, the nerve-injury-induced increased intracellular Ca
2+, and activation of the NR2B receptor [
42,
44]. Moreover, inhibiting Wnt3a/β-catenin signaling with the Wnt inhibitor IWP-2 reduced CCI-induced neuropathic pain, inhibiting synaptic plasticity in the spinal cord [
45]. These results highlight the contributions of neuronal Wnt signaling to the CCI-induced neuropathic pain via both canonical and non-canonical pathways.
Neuronal Wnt signaling in the spinal cord has an important role in HIV-induced neuralgia. Several Wnt ligands and β-catenin are upregulated in the spinal cord of HIV patients that experience chronic pain, but not in the pain-free ones [
46]. The injection of the viral protein HIV1-gp120, a model of neuropathic pain associated with HIV infection, induces Wnt3a upregulation in microglia [
47,
48], Wnt5a, and pro-inflammatory molecules IL-1β, IL-6, and TNF-α at the spinal cord level; intrathecal injection of Wnt5a antagonist Box5 significantly reduces the levels of inflammatory cytokines [
46]. Furthermore, the recombinant protein gp120 activates neurons by directly stimulating their NMDARs [
49,
50], leading to the synthesis and secretion of Wnt5a [
51]. Indeed, NMDAR is a key mediator of Wnt5a [
52], which plays a critical role in the differentiation and plasticity of excitatory synapses [
53,
54]. Recently, it was shown that Wnt5a could mediate HIV-related pain also via the Ror2/MMP2/IL-1β pathway [
55].
In addition, to directly modulate neuronal excitability and synaptic plasticity, Wnt signaling can induce neuroinflammation and recruit glial cells (see below).
3. Wnt Signaling Pathway in Glial Cells in Neuropathic Pain
Neuron–glia crosstalk through Wnt signaling may play a key role in the pathogenesis of several diseases, such as neurodegenerative conditions and chronic pain. Glial cells respond to secreted Wnt ligands which induce pro-inflammatory activation of glial cells, characterized by morphological changes and the release of pro-inflammatory mediators. The glial response, in turn, modulates neuronal function and the plasticity of neural circuits [
38]. Interestingly, regardless of the type of tissue involved or the kind of injury, the endogenous β-catenin-dependent Wnt signaling pathway is frequently activated at the site of tissue damage [
56] or along the pain pathway. Despite the experimental evidence is pointing out a significant role of the Wnt family in the physiological and pathological functioning of the spinal cord, cell-type-specific information is still lacking [
57,
58].
3.1. Astrocytes
Emerging evidence supports the role of canonical and non-canonical Wnt pathways as activators of astrocytes [
59,
60]. Under physiological conditions, astrocytes express a large panel of Wnt-related proteins. Astroglia are assumed to be the main source of Wnt ligands in the spinal cord, and harboring a wide variety of Wnt receptors, they are considered the main actor in the multidirectional astrocyte-neuron-microglia crosstalk [
61,
62]. Depending on the kind of injury and on the metabolic state of astroglia when activated, Wnt pathways modulate cell proliferation, glutamate uptake, the expression of glutamate transporters, pro-inflammatory cytokines, trophic factors, potassium, and water channels [
44,
62,
63,
64,
65,
66,
67].
Under physiological conditions, Wnt receptors show cell- and spatial-specific expression patterns [
68] that are altered after injury, indicating different cell-specific physiological roles at the spinal level [
58]. For example, after CCI, Fzd1 is transiently upregulated in spinal neurons, whereas Fzd8 is persistently upregulated in spinal astrocytes and satellite cells in DRGs [
29]. Furthermore, in the SNL model, Ryk is overexpressed on unmyelinated fibers, promoting the production and release of chemokine (C-C motif) ligand 2 (CCL2), which activates microglia [
44]. CCI-induced nociceptive hypersensitivity is significantly attenuated by hyperbaric oxygen treatment via suppressing the spinal kindlin-1/Wnt10a signaling pathway and activation of astrocytes [
69]. Kindlin-1 is a β-integrin binding protein that participates in the induction of inflammation and pain sensitization [
70,
71]. In CCI-treated rats, kindlin-1 is shown to be upregulated in spinal astrocytes [
70]. In accordance, downregulation of kindlin-1 reduces mechanical allodynia and astrocytic activation [
72]. This effect could be mediated by the modulation of Wnt expression by kindlin-1, as demonstrated in keratinocytes [
73]. Interestingly, the analgesic effect of dexmedetomidine, an agonist of α2-adrenergic receptors, used to treat a refractory form of neuropathic pain, administrated at late time points in the STZ-induced diabetic neuropathic pain model, is mediated by inhibition of the Wnt10a/β-catenin signaling pathway and astrocytic activation [
74]. Moreover, early-time-point dexmedetomidine administration relieves mechanical and thermal hyperalgesia by impeding microglial activation [
75].
In the CCI model, Ryk is upregulated in astrocytes and microglia in the SDH, and in satellite cells in DRGs [
42]. Wnt5a, Ryk, and ROR2 are overexpressed in different pain models, such as SNL, hind-paw injection of capsaicin, and HIV1-gp120 intrathecal injection [
76]. Importantly, in a rat neuropathic model of chronic post-thoracotomy pain (CPTP) and in other mouse models of neuropathic and inflammatory pain, the specific Wnt5a antagonist Box5 considerably inhibits the activation of astrocytes in the spinal cord and relieves mechanical allodynia and thermal hyperalgesia [
37,
77,
78,
79]. In the CPTP model, Liu and colleagues found that Ror2 predominantly co-localizes with astrocytes and modulates their activation, leading to a pro-inflammatory phenotype named A1 [
55]. Indeed, the knockdown of Ror2 promoted the neuroprotective phenotype of astrocytes (A2) versus the toxic one (A1) and attenuates mechanical hyperalgesia and thermal allodynia. Interestingly, Ror2 downregulation reduces the expression of C3aR in spinal astrocytes, suggesting that the modulatory effect of Ror2 on astrocytes phenotype can be mediated via C3aR expression [
55]. Wnt5a is upregulated only in pain-positive HIV1 patients [
46], whereas recombinant HIV1-gp120 induces Wnt5a neuronal release in an activity-dependent manner, causing hyperactivation of neurons [
80] and astrocytes [
81]. The HIV1-gp120-induced astrogliosis is sustained by the neuron to astrocyte Wnt5a-Ror2 signaling, and it is essential for HIV-associated pain sensitization [
81]. Both neuronal Wnt5a knockdown and astrocytic Ror2 knockdown abolish HIV1-gp120-induced astrogliosis and mechanical hyperalgesia [
81].
Taken together, these results indicate that activation of Wnt signaling contributes to the activation of astrocytes in the spinal cord, leading to neuroinflammation and chronic pain.
3.2. Microglia
Primary microglial cells and microglia-like cell lines respond to recombinant Wnt3a or Wnt5a application thanks to the localization on their membranes of a variety of Wnt receptors. Wnt3a and Wnt5a induce increased synthesis of pro-inflammatory molecules such as cytokines, chemokines, and cyclooxygenase 2 (COX2), and exacerbates the release of
de novo synthesized IL-6, IL-12, and TNFα [
82,
83], leading to neuroinflammation [
84]. Interestingly, when applied to cultured lipopolysaccharide (LPS)-primed microglial cells, recombinant Wnt3a and Wnt5a prompt dose-dependent downregulation of IL-6, COX-2, and TNFα expression, supporting a dual role of microglia as a pro or anti-inflammatory player, depending on the surrounding environment [
85,
86]. Furthermore, Wnt3a applied on primary microglial cells can induce exosome secretion, without inducing a neurotoxic pro-inflammatory phenotype [
87], underlining the major plasticity of microglia in responding to Wnt ligands.
Although Wnt pathways can prevent microglial activation and alleviate neuroinflammation [
84,
88,
89,
90], most studies indicate the involvement of Wnt pathways in the polarization of microglia toward a pro-inflammatory phenotype [
86,
91,
92,
93].
In several pain models in rodents, Wnt pathways induction activates microglial cells through different molecules: fractalkine (FKN) and BDNF, among others. In the HIV-1 gp120-induced pain model, activated microglia mediate synaptic degeneration. Interestingly, HIV infection prompts an increase in FKN [
94,
95,
96], a neuronal protein that regulates microglia-dependent synaptic phagocytosis. Since FKN is mainly expressed by neurons, and its CX3C chemokine receptor 1 (CX3CR1) is specifically present in microglia, the FKN pathway establishes signaling between neurons and microglia that leads to the regulation of synaptic pruning [
97,
98]. Recently, it has been shown that the HIV1-gp120 protein leads to the upregulation and release of Wnt3a in an NMDAR activity-dependent manner, resulting in activation of the β-catenin pathway and induction of FKN transcription in neurons [
99], ultimately resulting in synaptic degeneration of the neural spinal pain circuit. Furthermore, NMDAR antagonist DL-2-amino-5-phosphonovaleric acid (APV), the endogenous Wnt antagonist dickkopf-related protein 1 (DKK1), and knockout of CX3CR1, alleviate HIV1-gp120-induced mechanical allodynia in mice, suggesting a critical contribution of the Wnt/β-catenin/FKN/CX3CR1 pathway to HIV1-gp120-induced pain [
99]. Moreover, HIV1-gp120-induced neuropathic pain is mediated by the microglial release of BDNF, a crucial neuromodulator of pain transmission [
100]. Indeed, Wnt inhibitors block HIV1-gp120-induced BDNF release and subsequent induction of chronic pain, supporting a strong contribution of the Wnt pathway to spinal microglia activation, BDNF release, and chronic pain [
101,
102]. In a chemotherapy-induced neuropathic pain model, DKK1 significantly reduces capsaicin-induced inflammatory pain by blocking BDNF release from microglia, whereas the tankyrase inhibitor IWR-1-endo attenuates mechanical hyperalgesia [
103], inhibiting the activation of astrocytes, microglia, and TNF-α, and CCL2 and MAPK/ERK signaling in the spinal cord [
103].
Blocking Wnt signaling shows amelioration of neuropathic pain in other rodent pain models too. In fact, Zhang et al. showed that intrathecal injection of a Wnt signaling inhibitor, IWP-2, strongly diminishes both mechanical and thermal sensitization in CCI-operated rats via suppressing microglial reaction in the spinal cord [
29]. Meanwhile, targeting the Wnt/β-catenin signaling pathway with a tankyrase inhibitor XAV-939 suppresses the activation of microglia in the spinal cord and alleviates mechanical hypersensitivity in rats that undergo partial sciatic nerve ligation (pSNL) [
104]. The inhibition of β-catenin-independent Wnt pathways has recently been shown to reduce chronic pain via acting on microglia. For example, in a model of adjuvant-induced arthritis (AIA), the flavonoid crocin alleviates neuropathic pain by targeting Wnt5a signaling and microglia activation [
105]. Interestingly, astrocytes in the adult mouse brain express high levels of Wnt5a, which could serve as a novel astroglia–microglia communication pathway to be targeted in chronic pain conditions.
Recently, it was shown that the activation of the receptor complex DAP12-TREM2 contributes to the development of neuropathic pain. DAP12 signaling is triggered after nerve injury, whereas the direct activation of TREM2 induces mechanical allodynia in naïve mice. Moreover, DAP12-deficient mice fail to develop allodynia after nerve injury [
106]. DAP12 forms a receptor complex with TREM2 on the microglial membrane. Activation of this complex is associated with many physiological functions of microglia, and pathological conditions such as neurodegenerative diseases or chronic pain (summarized in [
107,
108]). Furthermore, the DAP12-TREM2 complex is involved in the survival of microglial cells via activation of Wnt/β-catenin signaling [
106,
109,
110].
Despite both Wnt signaling and microgliosis mediating maladaptive processes such as chronic pain, they are necessary for positive effects such as adult neurogenesis and synaptic plasticity [
111]. Thus, strict control of the balance between activation and inhibition of these phenomena is necessary for the maintenance of tissue homeostasis and proper physiological functions of the body. Interestingly, a recent paper demonstrated the need for early inflammation to reduce the risk of developing chronic pain later. The use of steroids or non-steroidal anti-inflammatory drugs (NSAIDs) and neutrophil depletion delayed the resolution of pain in animal models [
112]. Therefore, correct balance and timing between activation and inhibition of certain cell types are necessary for a positive physiological outcome.
This entry is adapted from the peer-reviewed paper 10.3390/cells11193143