Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Metabolic (dysfunction) associated fatty liver disease (MAFLD) is one of the most prevalent liver diseases and has no approved therapeutics. The high failure rates witnessed in late-phase MAFLD drug trials reflect the complexity of the disease, and how the disease develops and progresses remains to be fully understood. In vitro, human disease models play a pivotal role in mechanistic studies to unravel novel disease drivers and in drug testing studies to evaluate human-specific responses.
- MAFLD
- human model
- cell culture
- organoid
1. Introduction
1.1. MAFLD Epidemiology
MAFLD is a chronic liver disease composed of a spectrum of liver pathology primarily driven by the accumulation of fats in the tissue [1]. MAFLD patients are categorized by disease severity, including steatosis with no signs of liver injury, non-alcoholic steatohepatitis (NASH) with detectable levels of liver injury and various degrees of fibrosis, and end-stage cirrhosis [2]. MAFLD patients are often associated with a high risk for various metabolic diseases, such as type II diabetes mellitus (T2DM), cardiovascular disease (CVD), and chronic kidney disease (CKD) [3][4]. The condition is also widely considered a hepatic manifestation of metabolic syndromes [5]. MAFLD is proposed to be a more accurate nomenclature than ‘NAFLD’ given the disease’s inextricable link with metabolic syndromes [6]. Researchers use MAFLD when referring to all previous NAFLD studies. Globally, 1 out of 4 adults and 3–10% of children and adolescents develop MAFLD, and numbers will likely increase to 31% in adults by 2030 [7][8][9]. Moreover, in 2015, an estimated 20% of all MAFLD patients developed NASH, and the cases will likely increase to 27% by 2030 [9]. Patients with MAFLD and more severe liver fibrosis (stages F3 and F4 or cirrhosis) have an estimated higher risk of liver-related complications and deaths compared with the control group (fibrosis stages F0 to F2) [5][10]. The high incidence of MAFLD supports a pressing need for treating this metabolic disorder. However, no approved drug treatment is available for MAFLD patients [11][12]. Given the close pathophysiological link between MAFLD and type 2 diabetes (T2DM), drug development companies are actively exploring T2DM therapeutics such as thiazolidinediones, GLP1R glucagon-like peptide 1 receptor (GLP1R) agonists, farnesoid X receptor (FXR) agonists, and sodium–glucose cotransporter-2 (SGLT2) inhibitors for MAFLD treatment in ongoing human clinical trials [11]. Among the challenges faced in MAFLD drug discovery, developing physiologically relevant human models for mechanistic studies and screening of therapeutics remains a significant hurdle.
1.2. Genetic and Environmental Factors Driving MAFLD
The growing numbers of MAFLD-related studies have identified genetic and environmental factors responsible for disease development and progression. The increased risk factors observed in first-degree relatives and monozygotic twins support the heritability of MAFLD [13][14]. Genome-wide association studies (GWAS) in multiple NAFLD cohorts have identified risk variants in genes, including patatin-like phospholipase domain-containing 3 (PNPLA3), membrane-bound O-acyltransferase domain-containing 7 (MBOAT7), transmembrane 6 superfamily member 2 (TM6SF2), apolipoprotein C3 (APOC3), and glucokinase regulator GCKR [15][16]. Conversely, a protective single-nucleotide polymorphism (SNP) variant on the HSD17B13 gene locus was identified in an independent cohort [17]. This gene encodes for a poorly characterized enzyme (17-beta hydroxysteroid dehydrogenase 13) in the hepatocytes, and the reported mutation generates a truncated enzyme. Mechanistic insights on how this variant confers protective functions for MAFLD will likely reveal novel therapeutic targets. While population genetic studies across the globe identify more disease-associated mutations, much remains to be uncovered on how these reported genetic risk variants function. PNPLA3 is one of the earliest and most widely reported SNP variants identified in multiple independent cohorts, and how the encoded enzyme contributes to elevated MAFLD risk remains unclear. Conflicting reports of PNPLA3 contribution to hepatic steatosis in human in vitro models [18][19] and mouse in vivo models [20][21] highlighted the potential need to refine current human MAFLD models.
Similar to other metabolic syndromes and diseases, non-genetic drivers of MAFLD development and progression include sedentary lifestyles and nutrient-excessive diets. A high intake of fats and carbohydrates and a lack of physical activity promotes the excessive accumulation of lipids in the liver [22][23][24]. Notably, fructose intake correlated with fibrosis severity in MAFLD patients [23]. In comparison to glucose, fructose uptake and metabolism in the hepatocytes are relatively unregulated and, in the process, generate up to 100-fold more reactive oxygen species (ROS) production in the cells [25]. Besides being a substrate and inducer of de novo lipogenesis (DNL), fructose increases ER stress in the hepatocytes and stimulates pro-inflammatory response. A ‘two-hit’ or ‘multiple-hit’ hypothesis has been proposed for steatosis progression to NASH, where the buildup of fats in the hepatocytes prime the cells to inflammatory events stimulated by other agents such as bacterial toxins or metabolites [26][27]. Besides the high levels of nutrients supplemented in the MAFLD-associated ‘Western diet,’ the variety of food intake influences the gut microbiome, which strongly correlates with MAFLD progression and severity [28][29][30]. Metagenomic profiling of MAFLD patients at different stages by Loomba and colleagues identified microbial features uniquely enriched in MAFLD patients with advanced fibrosis [28]. In an independent study, Caussy et al. reported a similar phenomenon in MAFLD patients with cirrhosis [30]. To further understand how the microbiome plays a role in MAFLD development, Hoyles et al. performed a multi-omics study including metagenomics and phenomics profiling in obese patients with and without MAFLD [29]. The authors reported that the guts of obese individuals with early-stage MAFLD are low in microbe diversity and enriched for specific bacteria phyla such as Actinobacteria and Proteobacteria. Pathway analysis further identified corresponding increases in branched-chain amino acid (BCAA) and aromatic amino acid (AAA) metabolism [29]. Notably, the author demonstrated that phenylacetic acid (PAA), a microbial metabolic product of phenylalanine (an AAA), is enriched in the serum of the obese patient with steatosis, and PAA treatment induces MAFLD development in human hepatocytes and mice [29].
1.3. Molecular and Cellular Features of MAFLD Progression
The liver is composed of multiple cell types, including the parenchymal hepatocytes and cholangiocytes, as well as the non-parenchymal cells (NPCs) such as the liver sinusoidal endothelial cells (LSECs), hepatic stellate cells (HSCs), fibroblasts, and Kupffer cells (Figure 1) [31]. The cells are spatially arranged in the liver sinusoids to form networks of fluid channels such as the sinusoidal capillary and bile canaliculi. These various cell types and tissue structures are affected as MAFLD develops and are involved in disease progression at different stages [2]. The initial phase of MAFLD, hepatic steatosis, is defined by fat infiltration in more than 5% of the hepatocytes in the liver with no significant liver injury and fibrosis detected [5]. The flux of excessive non-esterified fatty acids (NEFAs) into the liver results in the visible accumulation of lipid vesicles of different sizes in the hepatocytes (Figure 2) [32][33]. The increased lipid species in the steatotic hepatocyte also reduces insulin sensitivity, a prominent molecular dysregulation in MAFLD progression [34][35]. Evidence from pre-clinical and clinical studies has shown that increased diacylglycerol (DAG) in hepatocytes upregulates protein kinase-Cε (PKCε) activity [36][37], which in turn inhibits the insulin signaling pathway by reducing phosphorylation of insulin receptor substrate-2 (IRS2) and phosphatidylinositol-3-OH kinase PI(3)K [38]. Insulin resistance in the cell, in turn, drives steatosis development by promoting DNL through increased glucokinase activity and impaired glycogen synthase activation. During disease progression, oxidation of the excess fatty acids increases ROS generation [39], and reduced glucose supply can further aggravate the process [40]. The increased ROS species promotes lipid peroxidation, resulting in the injury of organelles and the cellular membrane, and promotes apoptosis.
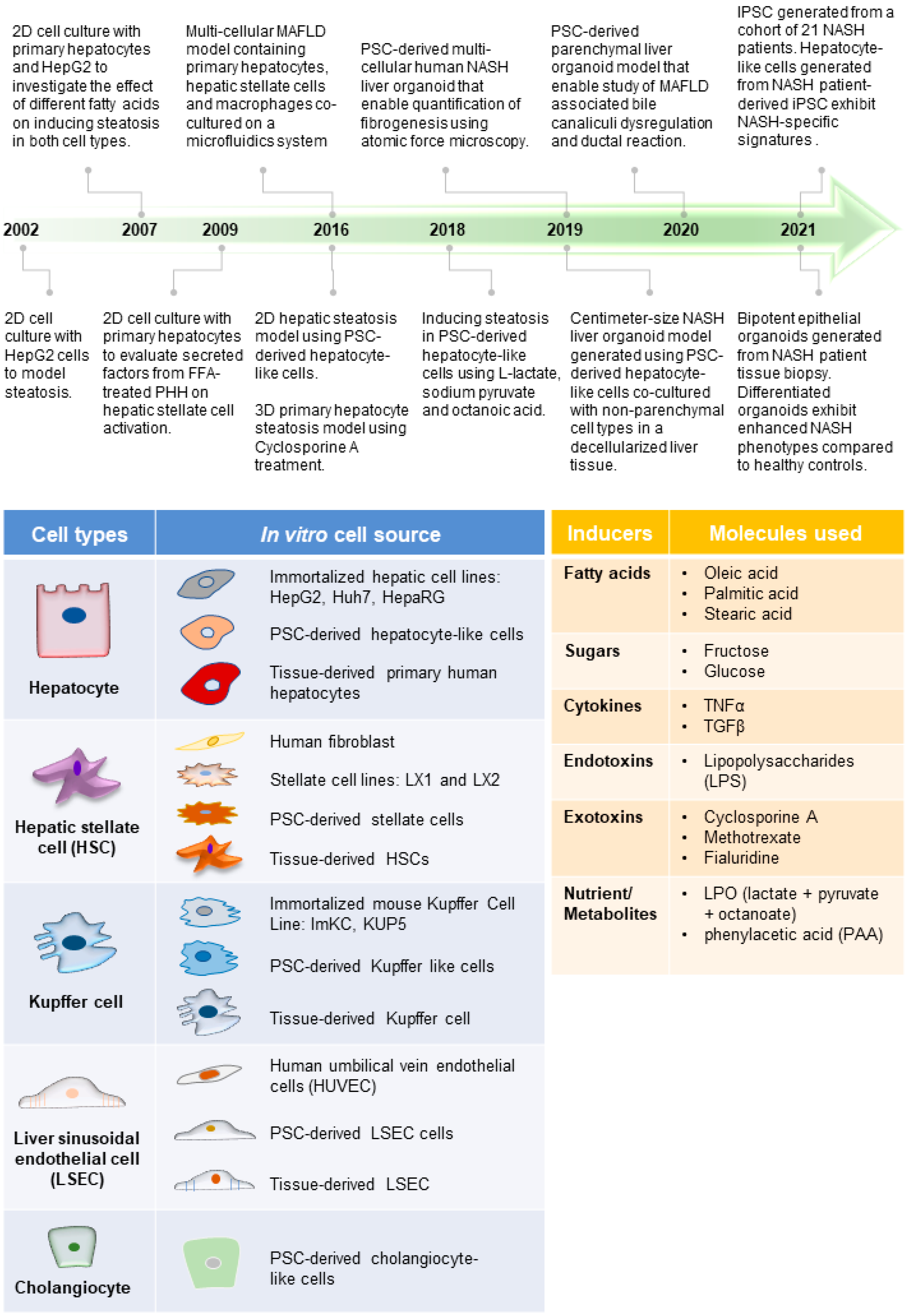
Figure 1. Advances in MAFLD human models. (Top) Timeline of representative MAFLD human model studies [41][42][43][44][45][46][47][48][49][50][51][52], which reflect how different cell types and the inducers of MAFLD (tables below) have been employed to create increasingly complex model systems. This includes the use of various cell culture platforms such as organoid culture systems as well as microfluidics. (Bottom) Tables listing the commonly used cell types for modeling different liver cells and molecules used in various studies to induce MAFLD phenotype (inducers).
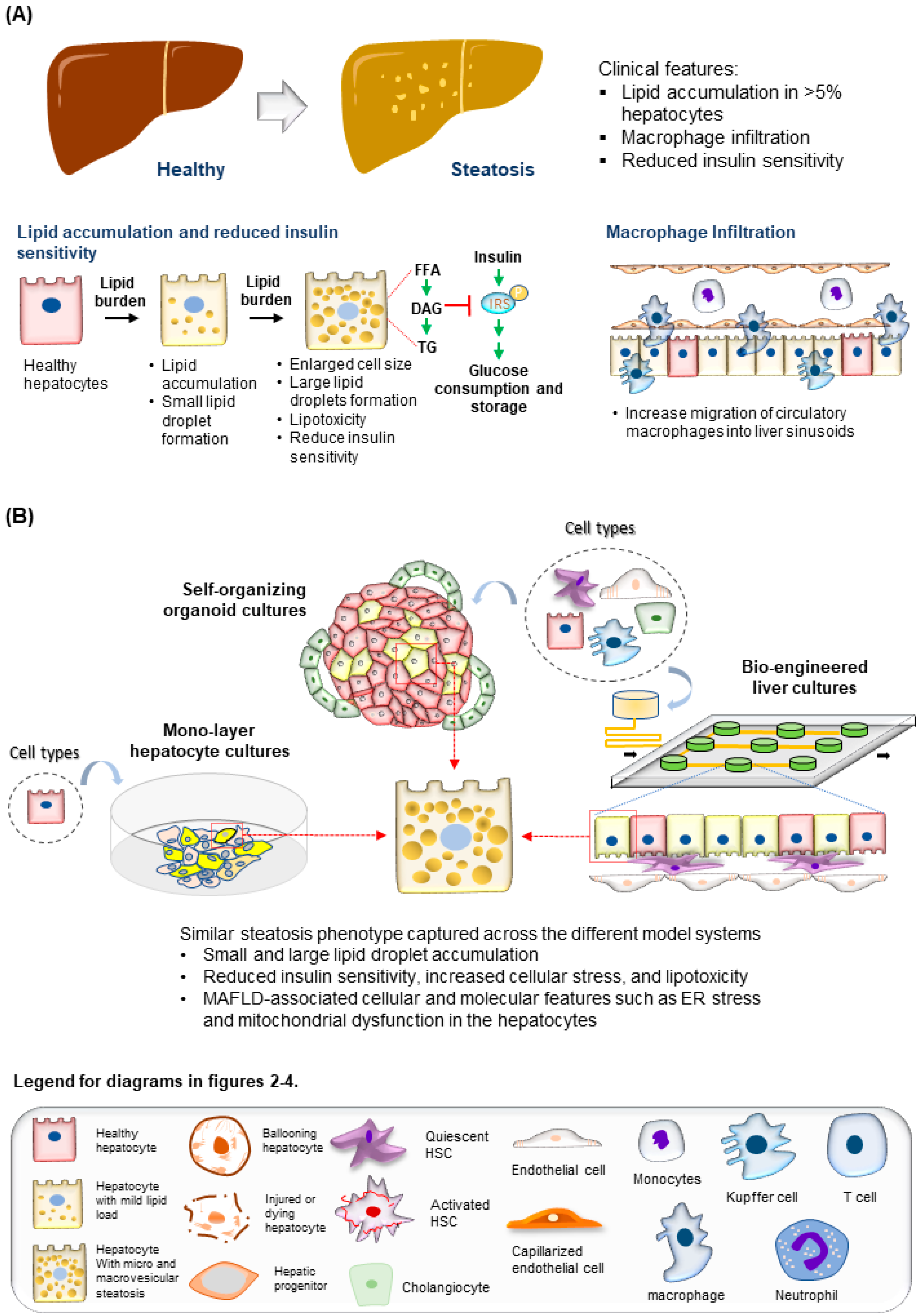
Figure 2. Modeling steatosis development. (A) (Top) Tissue features observed in liver biopsies from patients with steatosis. (Bottom) The major molecular and cellular changes during steatosis development in the liver. (B) (Top) Steatosis phenotypes observed in the hepatocytes are well recapitulated using various culture systems discussed. (Bottom) Legend for diagrams in Figure 2, Figure 3 and Figure 4.
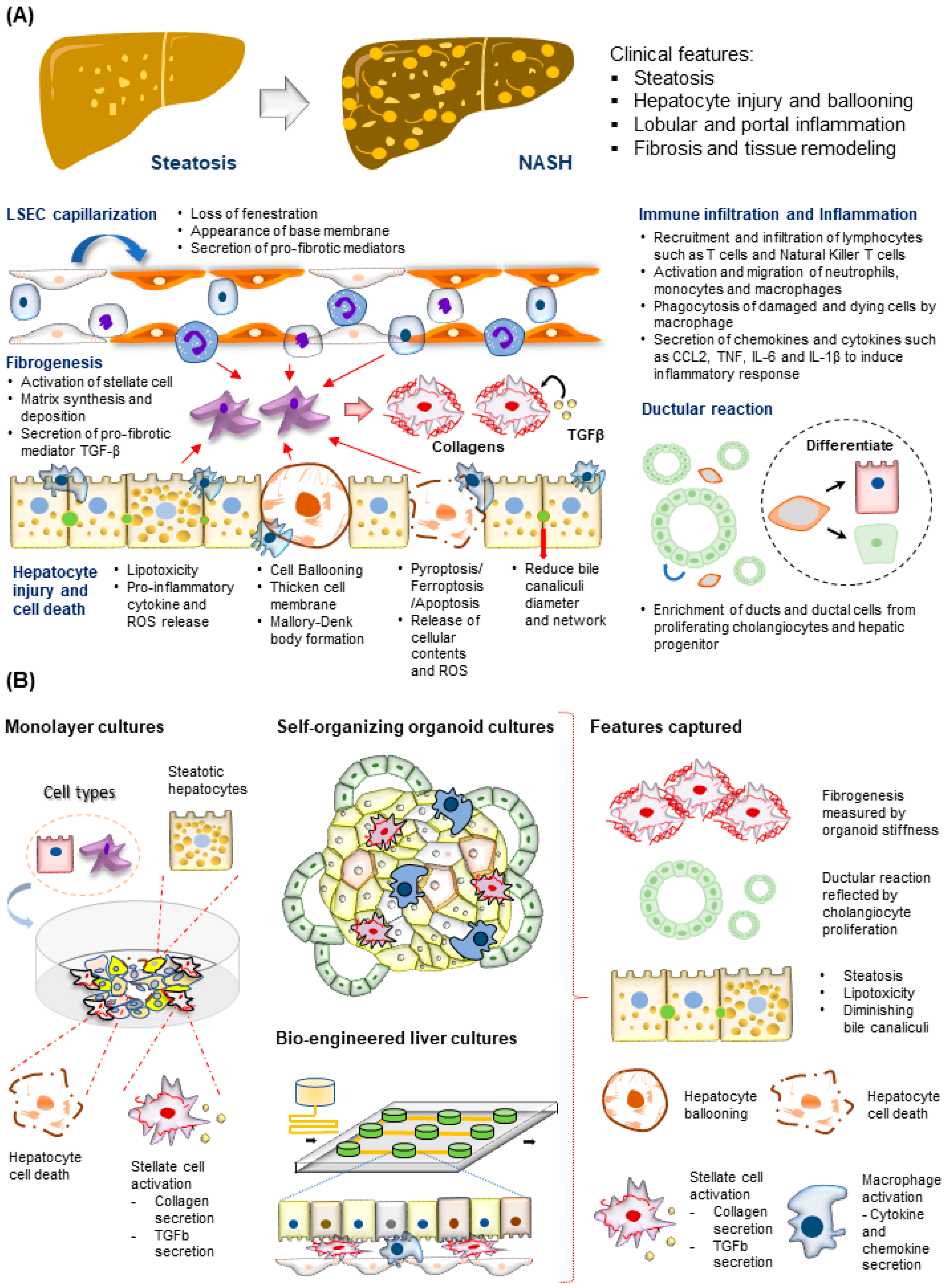
Figure 3. Modeling NASH development. (A) (Top) Tissue features observed in liver biopsies from patients with NASH. (Bottom) The major molecular and cellular changes during NASH development in the liver. This includes hepatocyte injury and cell death, immune cell infiltration and inflammation, fibrogenesis, LSEC capillarization, and ductular reaction. (B) (Left) NASH-associated cellular changes captured using monolayer cultures of hepatocytes and HSC. (Right) NASH-associated cellular and structural changes observed in the hepatocytes recapitulated using various 3D culture systems described.
The time taken for progression from steatosis to NASH varies among individuals and could take up to 14 years [53]. This lengthy period supports a dynamic interplay of interdependent inflammation and fibrogenesis activities, resulting in increased hepatocyte injury, cell death, and liver niche remodeling (Figure 3A). Other cell types involved in this process include both the innate and adaptive immune cells, such as macrophages, neutrophils, and T cells [54][55], and stromal cells, such as LSECs [56] and HSCs [57]. Significant drivers of inflammatory events include microbial pathogen-associated molecular patterns (PAMPs) from the gut, death-associated molecular patterns (DAMPs) released from apoptotic hepatocytes, immune-cell-released chemokines such as CXCL2 and CXCL5, and cytokines including TNFα, IL6, and IL10 [58][59][60]. The portal infiltration of immune cells such as macrophages can already be detected in the biopsy of the steatotic liver, supporting the involvement of immune cells at the early stages of disease progression [54]. A converging downstream effect of these inflammatory responses is the activation of the quiescent HSC, which plays a central role in remodeling the extra-cellular matrix (ECM) to promote fibrogenesis (Figure 3A) [61]. Major events include the PDGF-mediated HSC proliferation and migration to injury sites and TGFβ stimulation of HSC secretion of fibril-associated collagens and matrix metalloproteinase (MMP). These activities gradually remodel the soft and perforated endothelial/epithelial basal lamina to a stiff and obscure one rich in fibril-forming collagen types I and III. As fibrogenesis progress in NASH, the collagen fibrils thicken and form vast networks and septa that increasingly disrupt normal liver functions. Fibrogenesis remains the only histological feature predictive of clinical outcomes in NASH patients [62]. Besides inflammation and fibrogenesis, hepatocyte injury assessed by ‘hepatocellular ballooning’ is a defining pathological feature indicating disease progression from NAFLD to NASH (Figure 3A) [63]. These ‘ballooned’ hepatocytes have an enlarged cell size with a thickened cell membrane, rarefied cytoplasm, and the presence of the Mallory–Denk body [64][65]. Recapitulating these molecular and cellular features is essential for creating physiologically relevant human MAFLD models.
1.4. In Vitro Human Models for MAFLD
Animal models of MAFLD have been widely reported and used in drug evaluation studies [22][66]; however, significant species-specific differences between human and animal livers potentially hindered the clinical translation of discoveries generated from animal models [67][68]. The development of human in vitro MAFLD models is critical to providing a parallel platform to investigate human cells’ specific mechanisms and treatment responses. Over the past two decades, significant progress in creating human models of MAFLD in a dish has been achieved (Figure 1).
Researchers have engaged the use of increasingly complex culture systems to recapitulate different pathophysiological features in various stages of MAFLD. The models can be broadly classified into simple monolayer cultures composed mainly of single cell types (Table 1) or complex 3-dimensional (3D) cultures consisting of single or multiple liver cell types (Table 2). Various cell sources mimicking endogenous liver parenchymal cells and NPCs and inducers of lipid accumulation, including nutrients and metabolites, have been employed [29][41][42][69][70][71] (Figure 1). Monolayer cultures of human primary hepatocytes or immortalized cell lines provide a simple approach to model early hepatic cellular response under excessive free fatty acid (FFA) exposure (Table 1 and Figure 2B). However, MAFLD progression involves multiple cell type interactions and remodeling of the tissue environment [61]. Modeling such events during MAFLD progression requires more sophisticated cultures containing various liver cell types and the recapitulation of the cellular spatial organization (Figure 3A,B). Recent progress in bioengineering approaches and 3D cell culture techniques have further enabled the creation of such sophisticated models (Table 2).
Table 1. Human primary and immortalized hepatic cells used for modelling MAFLD.
| Cell Type | Cell Lines | Major Features |
|---|---|---|
| Immortalized Cell lines | HepG2 |
|
| Huh7 |
|
|
| WRL68 |
|
|
| HepaRG | ||
| Tissue-derived primary cells | Primary Hepatocyte (PHH) |
|
| Bipotent ductal stem cells |
|
|
| Pluripotent stem cells (PSC)-derived primary cells | Hepatocyte-like cells (HLC) |
|
2. Human Immortalized and Primary Cell Lines for Modeling MAFLD
Human hepatic cells widely employed in MAFLD studies include immortalized hepatocytes, cancer cell lines, primary hepatocytes, and pluripotent stem cell (PSC)-derived hepatocyte-like cells (HLCs) (Table 1). The monolayer culture of these cell lines is primarily used to model hepatocyte uptake of lipids, including the transport, storage, and metabolism of the excess lipids. In addition, the homogenous cell culture is suitable for dissecting direct molecular and cellular responses of hepatocytes exposed to nutrition and environmental changes in the liver through manipulating the cell culture media. The stable cell lines also allow further genetic perturbations for gain and loss of function studies.
2.1. Immortalized Hepatic Cell Lines for Modeling MAFLD
One of the most widely used hepatic cell lines for modeling human MAFLD is the HepG2 cell line derived from hepatocellular carcinoma (HCC) and subsequently immortalized for stable culture in vitro [85]. HepG2 cells express and secrete a variety of major hepatic plasma proteins, including albumin, and exhibit several hepatocyte-specific responses to environmental stimuli, making the cells suitable for in vitro modeling of hepatocyte functions [86][87]. Several treatment regimens and detection assays have been widely adopted to induce and measure steatosis in HepG2 cells. Treatment of HepG2 cells with oleic acid (OA), a monounsaturated fatty acid, could cause the accumulation of intracellular triglyceride and lipid droplets within the cells. These phenotypic changes can be easily detected by staining with lipid dyes such as Oil red or Nile red and quantified with lipid assay kits [43][44][72]. On the other hand, OA treatment is insufficient to induce changes in the cellular oxidative phosphorylation state, which was dysregulated in NASH patients [88]. This phenotype was recapitulated by co-treatment with saturated fatty acids, such as palmitic acid (PA) or stearic acid (SA) [73][74]. HepG2 cells treated with different ratios of OA to PA develop varying steatosis phenotypes; a higher proportion of OA results in benign chronic steatosis, while a higher PA proportion induces more significant toxic and apoptotic effects in the cells [44]. In addition, PA treatment induces the production of pro-inflammatory chemokine IL-8 in HepG2 cells, recapitulating the elevated IL-8 levels seen in NASH patients [73][89][90]. This simple system is also employed for mechanistic MAFLD studies and the validation of therapeutics [75][91]. C1q/TNF-related protein 9 (CTRP9) is an adiponectin paralog expressed by adipose tissue, and overexpression of this protein has been shown to reduce steatosis in mouse models [92]. To further unravel how CTRP9 functions in the human hepatocyte, Jung et al. utilized PA-treated HepG2 cells which similarly exhibited reduced hepatic steatosis upon CTRP9 treatment. The authors unravel that CTRP9 induces protective effects against steatosis through the inhibition of ER stress via the activation of AMPK-mediated induction of autophagy [91].
A similar immortalized cell line of growing interest for MAFLD modeling is HepaRG [78][79]. These cells exhibit greater sensitivity to drug-induced steatosis [78] and features of MAFLD upon treatment with free fatty acids [79]. Other immortalized hepatic cell lines, such as fetal liver-derived WRL-68 and HCC-derived Huh7 cells, are also used in MAFLD modeling (Table 1) [69][76][77]. All these immortalized cell lines have provided a simple approach to modeling the disease, and the ease of culture and high cell viability have made them excellent cell options for engineering approaches to create more complex cellular models. However, these cells may not fully reflect the primary hepatocyte due to the cancer origins of some of the cell lines and the cellular changes induced by the transgenic approaches used to immortalize the cell. The altered hepatic nature of the cells raised concerns about the accuracy of observations from MAFLD models generated with these cells [85][93].
2.2. Liver-Tissue-Derived Primary Cells for Modeling MAFLD
Primary human hepatocytes (PHHs) derived from liver tissue have been a source of physiologically relevant in vitro models for liver diseases, including MAFLD [41][44][45][70][73][80][81][91][94][95][96]. The PHH culture faithfully retains many molecular and cellular features of their in vivo counterparts and the disease phenotypes of the liver origin. Similar to HepG2 cells, PHHs respond differently to the varying ratios of OA and PA used in treatment [44]. The differential response to OA and PA was validated by analyzing the cell viability, gene expression, and secretome of treated cells [44][45][80][81][91][95][96]. Treatment of PHHs with PA induces triglyceride accumulation, elevated expression of lipogenic genes, a significant increase in IL-8 release, and increased ER stress [73][91][96][97][98]. In addition, BMP-8B was induced in OA-treated PHHs, correlating with observed upregulation in the human steatotic liver tissue [81]. PHH MAFLD models were also employed to evaluate therapeutic modalities such as CTRP9 and Humanin to alleviate the steatosis phenotype induced by FFA treatment [91][96]. However, the lack of self-renewing capacity and a limited number of cells that can be derived from a single liver poses significant challenges to using PHHs in large-scale applications. The limited PHH source also results in a high cost, further restricting their adoption in MAFLD studies. More importantly, the molecular and functional differences observed in hepatocytes from different individuals may influence the reproducibility of results. Prill et al. evaluated the FFA treatment response of hepatocytes derived from five donors where two donors had the reported TM6SF2 E167K genetic variant [82]. While the study validated the effects of the TM6SF2 E167K genetic variant in conferring susceptibility to steatosis, the results also highlight variability in FFA treatment response observed across different donor hepatocytes. Idiosyncratic responses have been a significant limitation for using these cells for liver toxicology and disease modeling [99].
To overcome the abovementioned limitations, scientists have attempted to isolate expandable primary hepatic progenitors from human liver biopsies [100][101][102]. Clever’s group first reported the successful expansion of bipotent cholangiocyte progenitors from human liver tissue, which can differentiate into functional hepatocytes in vitro [100]. The group subsequently optimized the protocol to achieve the culture of expandable human hepatocyte progenitors that can more efficiently generate hepatocytes with improved functions and engraftment efficacy [102]. In parallel, several groups have also reported the successful derivation of expandable hepatic progenitors from isolated human hepatocytes [103][104] and human fetal liver, respectively [105]. Similarly, these expandable progenitors are a renewable cell source for generating functional primary hepatocytes. In addition, as the cells can expand and maintain their progenitor state, transgenic cell lines can be generated with genome editing tools to facilitate the study of gene functions [106][107]. McCarron et al. employed the cholangiocyte progenitor culture method [100] and generated NASH patient-specific models from their liver biopsies [46]. The cells derived from NASH patient tissue exhibit significant dysregulation in lipid metabolism and hepatic functions. They also express pro-inflammatory and fibrogenesis-associated genes and have reduced proliferative capacity. Of concern, these observations are idiopathic and not captured across cell models from different NASH patients. Intriguingly, the authors also showed that the NASH patient-derived cells express high levels of Ubiquitin D (UBD), an inhibitor of RNA virus-induced interferon signaling, and hypothesized that they are more susceptible to virus infection. The authors demonstrated that the NASH patient-derived cell models can be more easily infected by SARS-CoV-2 pseudovirus (vesicular stomatitis virus (VSV) expressing SARS-CoV-2 spike protein), correlating with the reports of more severe COVID-19 infections in NASH patients [108][109]. Despite these breakthroughs, significant challenges exist for the wide adoption of this liver progenitor culture system. In the clinic, the invasive nature, high cost of liver biopsies, and limited drug treatment options dissuade MAFLD patients from undergoing the surgical procedure [110]. This limits the accessibility of patient liver tissue for establishing the cell cultures. There are also significant hurdles for each progenitor culture system [100][102]. The cholangiocyte progenitor culture system, which enables efficient derivation and long-term culture of cells from the NASH patient [100], is inefficient in generating functional hepatocytes [102]. On the other hand, the hepatocyte progenitor culture system, which efficiently generates highly functional hepatocytes, could only be stably derived from fetal tissues [102].
2.3. PSC-Derived Hepatic Cells for MAFLD Modeling
PSCs such as embryonic stem cells derived from blastocysts or induced pluripotent stem cells (iPSCs) generated from adult somatic cells are highly renewable cell sources with the capacity to differentiate into cell types of all three germ layers [111][112]. Harnessing the PSC’s ability to generate endoderm lineage cell types, including lung, gastrointestinal tract, liver, and pancreas, multiple groups have established protocols to derive functional hepatocytes and cholangiocytes from PSCs in vitro [113][114][115][116][117][118][119][120][121]. While these PSC-derived hepatocytes expressed cell-type-specific markers such as ALB, CK8, HNF4A, and CYP3A4, the PSC-derived cells are fetal in nature. The cells express high levels of alpha-fetoprotein (AFP), not found in healthy adult liver tissue. They also lack mature hepatocyte features and functions, including the absence of mature cytochrome proteins and response to drug treatment [122]. Hence, these cells are often referred to as HLCs. These PSC-derived HLCs can be utilized in various primary hepatocyte-related applications, from modeling liver development and diseases [47][114][123][124][125][126] to cell-based regenerative applications [127]. In modeling MAFLD, HLCs treated with FFAs could accumulate lipid droplets, exhibit cellular stress responses, express genes associated with lipid metabolism, and recapitulate transcriptional profiles similar to MAFLD liver tissues [19][47][48][49][83][84]. The self-renewing capacity of PSCs enables scalable production of HLCs for applications requiring a large number of cells. Harnessing this inherent advantage, Parafati et al. utilized PSC-derived HLCs in a 13,000-compound high-throughput screen to identify molecules that can reverse ER-stress-induced steatosis [83]. The study revealed that cyclin-dependent kinase (CDK) inhibitors can attenuate steatosis through the cyclin D3-CDK2-4/CCAAT-enhancer-binding proteins/diacylglycerol acyltransferase 2 pathway.
An inherent advantage of modeling disease using the iPSC platform is the generation of genetic models through genome editing of iPSC cell lines or the establishment of iPSC lines from patients with the genotype of interest [49][112][128][129]. To dissect the role of PNPLA3, the most widely reported gene associated with the full spectrum of MAFLD, Tilson et al. generated PNPLA3 knockout and PNPLA3 I148M variants in isogenic iPSC cell lines using the CRISPR genome editing tool [19]. In contrast to the mouse model with similar genetic edits, the human HLCs with depleted PNPLA3 and I148M variants demonstrated increased steatosis with significant improvements in cell viability under saturated fatty acid treatment. Furthermore, the authors revealed that PNPLA3 depletion and expression of the I148M variant in the cells increased polyunsaturated fats and altered lipid metabolism profiles, which correlated with patient tissue profiles. While these changes protected the cells from lipotoxicity, they also sensitized them to drug- and alcohol-induced injury. This study highlighted the main advantages of MAFLD studies with human cellular models, where genetic manipulations and biochemical assays with homogenous human hepatocyte cultures are advantageous for mechanistic studies. A parallel study by Duwaerts et al. further highlighted the iPSC platform’s potential for unraveling novel genetic drivers of MAFLD by generating patient-specific MAFLD models [49]. The team established 21 iPSC cell lines from MAFLD patients, including 18 NASH patients. Intriguingly, iPSC-HLCs generated from NASH patients had increased lipid droplet accumulation and expressed distinct transcriptomic profiles compared to iPSC-HLCs generated from individuals without MAFLD. Dysregulated genes are associated with cell death and transformation, insulin resistance, and cellular oxygen consumption. This observation supports the existence of genetic or epigenetic drivers of the MAFLD phenotype in the iPSC cell lines. Further in-depth genomics study and expansion of sample sizes would likely facilitate the identification of MAFLD regulatory genes and mutations. Importantly, this study underlines the potential of generating patient-specific MAFLD models for drug testing, establishing the foundation for future precision therapy.
This entry is adapted from the peer-reviewed paper 10.3390/ijms231911850
References
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344.
- Yeh, M.M.; Brunt, E.M. Pathological features of fatty liver disease. Gastroenterology 2014, 147, 754–764.
- Wang, T.Y.; Wang, R.F.; Bu, Z.Y.; Targher, G.; Byrne, C.D.; Sun, D.Q.; Zheng, M.H. Association of metabolic dysfunction-associated fatty liver disease with kidney disease. Nat. Rev. Nephrol. 2022, 18, 259–268.
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801.
- Hardy, T.; Oakley, F.; Anstee, Q.M.; Day, C.P. Nonalcoholic Fatty Liver Disease: Pathogenesis and Disease Spectrum. Annu. Rev. Pathol. 2016, 11, 451–496.
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1.
- Le Garf, S.; Negre, V.; Anty, R.; Gual, P. Metabolic Fatty Liver Disease in Children: A Growing Public Health Problem. Biomedicines 2021, 9, 1915.
- Lazarus, J.V.; Mark, H.E.; Villota-Rivas, M.; Palayew, A.; Carrieri, P.; Colombo, M.; Ekstedt, M.; Esmat, G.; George, J.; Marchesini, G.; et al. The global NAFLD policy review and preparedness index: Are countries ready to address this silent public health challenge? J. Hepatol. 2022, 76, 771–780.
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133.
- Sanyal, A.J.; Van Natta, M.L.; Clark, J.; Neuschwander-Tetri, B.A.; Diehl, A.; Dasarathy, S.; Loomba, R.; Chalasani, N.; Kowdley, K.; Hameed, B.; et al. Prospective Study of Outcomes in Adults with Nonalcoholic Fatty Liver Disease. N. Engl. J. Med. 2021, 385, 1559–1569.
- Ferguson, D.; Finck, B.N. Emerging therapeutic approaches for the treatment of NAFLD and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2021, 17, 484–495.
- Dufour, J.F.; Anstee, Q.M.; Bugianesi, E.; Harrison, S.; Loomba, R.; Paradis, V.; Tilg, H.; Wong, V.W.; Zelber-Sagi, S. Current therapies and new developments in NASH. Gut 2022, 71, 2123–2134.
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279.
- Loomba, R.; Schork, N.; Chen, C.H.; Bettencourt, R.; Bhatt, A.; Ang, B.; Nguyen, P.; Hernandez, C.; Richards, L.; Salotti, J.; et al. Heritability of Hepatic Fibrosis and Steatosis Based on a Prospective Twin Study. Gastroenterology 2015, 149, 1784–1793.
- Eslam, M.; George, J. Genetic contributions to NAFLD: Leveraging shared genetics to uncover systems biology. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 40–52.
- Anstee, Q.M.; Seth, D.; Day, C.P. Genetic Factors That Affect Risk of Alcoholic and Nonalcoholic Fatty Liver Disease. Gastroenterology 2016, 150, 1728–1744.e7.
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106.
- Huang, Y.; Cohen, J.C.; Hobbs, H.H. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J. Biol. Chem. 2011, 286, 37085–37093.
- Tilson, S.G.; Morell, C.M.; Lenaerts, A.S.; Park, S.B.; Hu, Z.; Jenkins, B.; Koulman, A.; Liang, T.J.; Vallier, L. Modeling PNPLA3-Associated NAFLD Using Human-Induced Pluripotent Stem Cells. Hepatology 2021, 74, 2998–3017.
- Basantani, M.K.; Sitnick, M.T.; Cai, L.; Brenner, D.S.; Gardner, N.P.; Li, J.Z.; Schoiswohl, G.; Yang, K.; Kumari, M.; Gross, R.W.; et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J. Lipid Res. 2011, 52, 318–329.
- Ochi, T.; Munekage, K.; Ono, M.; Higuchi, T.; Tsuda, M.; Hayashi, Y.; Okamoto, N.; Toda, K.; Sakamoto, S.; Oben, J.A.; et al. Patatin-like phospholipase domain-containing protein 3 is involved in hepatic fatty acid and triglyceride metabolism through X-box binding protein 1 and modulation of endoplasmic reticulum stress in mice. Hepatol. Res. 2016, 46, 584–592.
- Anstee, Q.M.; Goldin, R.D. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int. J. Exp. Pathol. 2006, 87, 1–16.
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 993–999.
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M.; Nonalcoholic Steatohepatitis Clinical Research Network. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1961–1971.
- Jegatheesan, P.; De Bandt, J.P. Fructose and NAFLD: The Multifaceted Aspects of Fructose Metabolism. Nutrients 2017, 9, 230.
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845.
- Tilg, H.; Adolph, T.E.; Moschen, A.R. Multiple Parallel Hits Hypothesis in Nonalcoholic Fatty Liver Disease: Revisited After a Decade. Hepatology 2021, 73, 833–842.
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e5.
- Hoyles, L.; Fernandez-Real, J.M.; Federici, M.; Serino, M.; Abbott, J.; Charpentier, J.; Heymes, C.; Luque, J.L.; Anthony, E.; Barton, R.H.; et al. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 2018, 24, 1070–1080.
- Caussy, C.; Tripathi, A.; Humphrey, G.; Bassirian, S.; Singh, S.; Faulkner, C.; Bettencourt, R.; Rizo, E.; Richards, L.; Xu, Z.Z.; et al. A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat. Commun. 2019, 10, 1406.
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1147–R1151.
- Tandra, S.; Yeh, M.M.; Brunt, E.M.; Vuppalanchi, R.; Cummings, O.W.; Ünalp-Arida, A.; Wilson, L.A.; Chalasani, N. Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease. J. Hepatol. 2011, 55, 654–659.
- Marrache, M.K.; Rockey, D.C. Statins for treatment of chronic liver disease. Curr. Opin. Gastroenterol. 2021, 37, 200–207.
- Marchesini, G.; Brizi, M.; Morselli-Labate, A.M.; Bianchi, G.; Bugianesi, E.; McCullough, A.J.; Forlani, G.; Melchionda, N. Association of nonalcoholic fatty liver disease with insulin resistance. Am. J. Med. 1999, 107, 450–455.
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91.
- Petersen, M.C.; Madiraju, A.K.; Gassaway, B.M.; Marcel, M.; Nasiri, A.R.; Butrico, G.; Marcucci, M.J.; Zhang, D.; Abulizi, A.; Zhang, X.M.; et al. Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J. Clin. Investig. 2016, 126, 4361–4371.
- Magkos, F.; Su, X.; Bradley, D.; Fabbrini, E.; Conte, C.; Eagon, J.C.; Varela, J.E.; Brunt, E.M.; Patterson, B.W.; Klein, S. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 2012, 142, 1444–1446.e2.
- Samuel, V.T.; Liu, Z.X.; Wang, A.; Beddow, S.A.; Geisler, J.G.; Kahn, M.; Zhang, X.M.; Monia, B.P.; Bhanot, S.; Shulman, G.I. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Investig. 2007, 117, 739–745.
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495.
- Assaily, W.; Rubinger, D.A.; Wheaton, K.; Lin, Y.; Ma, W.; Xuan, W.; Brown-Endres, L.; Tsuchihara, K.; Mak, T.W.; Benchimol, S. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol. Cell 2011, 44, 491–501.
- Bell, C.C.; Hendriks, D.F.; Moro, S.M.; Ellis, E.; Walsh, J.; Renblom, A.; Fredriksson Puigvert, L.; Dankers, A.C.; Jacobs, F.; Snoeys, J.; et al. Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease. Sci. Rep. 2016, 6, 25187.
- Lyall, M.J.; Cartier, J.; Thomson, J.P.; Cameron, K.; Meseguer-Ripolles, J.; O’Duibhir, E.; Szkolnicka, D.; Villarin, B.L.; Wang, Y.; Blanco, G.R.; et al. Modelling non-alcoholic fatty liver disease in human hepatocyte-like cells. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170362.
- Okamoto, Y.; Tanaka, S.; Haga, Y. Enhanced GLUT2 gene expression in an oleic acid-induced in vitro fatty liver model. Hepatol. Res. 2002, 23, 138–144.
- Gomez-Lechon, M.J.; Donato, M.T.; Martinez-Romero, A.; Jimenez, N.; Castell, J.V.; O’Connor, J.E. A human hepatocellular in vitro model to investigate steatosis. Chem. Biol. Interact. 2007, 165, 106–116.
- Wobser, H.; Dorn, C.; Weiss, T.S.; Amann, T.; Bollheimer, C.; Buttner, R.; Scholmerich, J.; Hellerbrand, C. Lipid accumulation in hepatocytes induces fibrogenic activation of hepatic stellate cells. Cell Res. 2009, 19, 996–1005.
- McCarron, S.; Bathon, B.; Conlon, D.M.; Abbey, D.; Rader, D.J.; Gawronski, K.; Brown, C.D.; Olthoff, K.M.; Shaked, A.; Raabe, T.D. Functional Characterization of Organoids Derived From Irreversibly Damaged Liver of Patients With NASH. Hepatology 2021, 74, 1825–1844.
- Ramli, M.N.B.; Lim, Y.S.; Koe, C.T.; Demircioglu, D.; Tng, W.; Gonzales, K.A.U.; Tan, C.P.; Szczerbinska, I.; Liang, H.; Soe, E.L.; et al. Human Pluripotent Stem Cell-Derived Organoids as Models of Liver Disease. Gastroenterology 2020, 159, 1471–1486.e12.
- Graffmann, N.; Ring, S.; Kawala, M.A.; Wruck, W.; Ncube, A.; Trompeter, H.I.; Adjaye, J. Modeling Nonalcoholic Fatty Liver Disease with Human Pluripotent Stem Cell-Derived Immature Hepatocyte-Like Cells Reveals Activation of PLIN2 and Confirms Regulatory Functions of Peroxisome Proliferator-Activated Receptor Alpha. Stem Cells Dev. 2016, 25, 1119–1133.
- Duwaerts, C.C.; Le Guillou, D.; Her, C.L.; Phillips, N.J.; Willenbring, H.; Mattis, A.N.; Maher, J.J. Induced Pluripotent Stem Cell-derived Hepatocytes From Patients With Nonalcoholic Fatty Liver Disease Display a Disease-specific Gene Expression Profile. Gastroenterology 2021, 160, 2591–2594.e6.
- Ouchi, R.; Togo, S.; Kimura, M.; Shinozawa, T.; Koido, M.; Koike, H.; Thompson, W.; Karns, R.A.; Mayhew, C.N.; McGrath, P.S.; et al. Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metab. 2019, 30, 374–384.e6.
- Wang, Y.; Wang, H.; Deng, P.; Tao, T.; Liu, H.; Wu, S.; Chen, W.; Qin, J. Modeling Human Nonalcoholic Fatty Liver Disease (NAFLD) with an Organoids-on-a-Chip System. ACS Biomater. Sci. Eng. 2020, 6, 5734–5743.
- Ratziu, V.; Sanyal, A.; Harrison, S.A.; Wong, V.W.; Francque, S.; Goodman, Z.; Aithal, G.P.; Kowdley, K.V.; Seyedkazemi, S.; Fischer, L.; et al. Cenicriviroc Treatment for Adults With Nonalcoholic Steatohepatitis and Fibrosis: Final Analysis of the Phase 2b CENTAUR Study. Hepatology 2020, 72, 892–905.
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654.e9; quiz e39–e40.
- Gadd, V.L.; Skoien, R.; Powell, E.E.; Fagan, K.J.; Winterford, C.; Horsfall, L.; Irvine, K.; Clouston, A.D. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014, 59, 1393–1405.
- Syn, W.K.; Oo, Y.H.; Pereira, T.A.; Karaca, G.F.; Jung, Y.; Omenetti, A.; Witek, R.P.; Choi, S.S.; Guy, C.D.; Fearing, C.M.; et al. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology 2010, 51, 1998–2007.
- Miyao, M.; Kotani, H.; Ishida, T.; Kawai, C.; Manabe, S.; Abiru, H.; Tamaki, K. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab. Investig. 2015, 95, 1130–1144.
- Schwabe, R.F.; Tabas, I.; Pajvani, U.B. Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology 2020, 158, 1913–1928.
- Alisi, A.; Carsetti, R.; Nobili, V. Pathogen- or damage-associated molecular patterns during nonalcoholic fatty liver disease development. Hepatology 2011, 54, 1500–1502.
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218.
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194.
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564.
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565.
- Matteoni, C.A.; Younossi, Z.M.; Gramlich, T.; Boparai, N.; Liu, Y.C.; McCullough, A.J. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999, 116, 1413–1419.
- Lackner, C.; Gogg-Kamerer, M.; Zatloukal, K.; Stumptner, C.; Brunt, E.M.; Denk, H. Ballooned hepatocytes in steatohepatitis: The value of keratin immunohistochemistry for diagnosis. J. Hepatol. 2008, 48, 821–828.
- Kucukoglu, O.; Guldiken, N.; Chen, Y.; Usachov, V.; El-Heliebi, A.; Haybaeck, J.; Denk, H.; Trautwein, C.; Strnad, P. High-fat diet triggers Mallory-Denk body formation through misfolding and crosslinking of excess keratin 8. Hepatology 2014, 60, 169–178.
- Bhattacharjee, J.; Kumar, J.M.; Arindkar, S.; Das, B.; Pramod, U.; Juyal, R.C.; Majumdar, S.S.; Nagarajan, P. Role of immunodeficient animal models in the development of fructose induced NAFLD. J. Nutr. Biochem. 2014, 25, 219–226.
- Takahashi, Y.; Soejima, Y.; Fukusato, T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2012, 18, 2300–2308.
- Teufel, A.; Itzel, T.; Erhart, W.; Brosch, M.; Wang, X.Y.; Kim, Y.O.; von Schonfels, W.; Herrmann, A.; Bruckner, S.; Stickel, F.; et al. Comparison of Gene Expression Patterns Between Mouse Models of Nonalcoholic Fatty Liver Disease and Liver Tissues From Patients. Gastroenterology 2016, 151, 513–525.e0.
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J. Gastroenterol. Hepatol. 2009, 24, 830–840.
- Piras, I.S.; Gerhard, G.S.; DiStefano, J.K. Palmitate and Fructose Interact to Induce Human Hepatocytes to Produce Pro-Fibrotic Transcriptional Responses in Hepatic Stellate Cells Exposed to Conditioned Media. Cell Physiol. Biochem. 2020, 54, 1068–1082.
- Todoric, J.; Di Caro, G.; Reibe, S.; Henstridge, D.C.; Green, C.R.; Vrbanac, A.; Ceteci, F.; Conche, C.; McNulty, R.; Shalapour, S.; et al. Fructose stimulated de novo lipogenesis is promoted by inflammation. Nat. Metab. 2020, 2, 1034–1045.
- Cui, W.; Chen, S.L.; Hu, K.Q. Quantification and mechanisms of oleic acid-induced steatosis in HepG2 cells. Am. J. Transl. Res. 2010, 2, 95–104.
- Joshi-Barve, S.; Barve, S.S.; Amancherla, K.; Gobejishvili, L.; Hill, D.; Cave, M.; Hote, P.; McClain, C.J. Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology 2007, 46, 823–830.
- García-Ruiz, I.; Solís-Muñoz, P.; Fernández-Moreira, D.; Muñoz-Yagüe, T.; Solís-Herruzo, J.A. In vitro treatment of HepG2 cells with saturated fatty acids reproduces mitochondrial dysfunction found in nonalcoholic steatohepatitis. Dis. Model. Mech. 2015, 8, 183–191.
- Khalifa, O.; Al-Akl, N.S.; Errafii, K.; Arredouani, A. Exendin-4 alleviates steatosis in an in vitro cell model by lowering FABP1 and FOXA1 expression via the Wnt/-catenin signaling pathway. Sci. Rep. 2022, 12, 2226.
- Gunn, P.J.; Green, C.J.; Pramfalk, C.; Hodson, L. In vitro cellular models of human hepatic fatty acid metabolism: Differences between Huh7 and HepG2 cell lines in human and fetal bovine culturing serum. Physiol. Rep. 2017, 5, e13532.
- Chen, K.Y.; Lin, J.A.; Yao, H.Y.; Hsu, A.C.; Tai, Y.T.; Chen, J.T.; Hsieh, M.C.; Shen, T.L.; Hsu, R.Y.; Wu, H.T.; et al. Arctigenin protects against steatosis in WRL68 hepatocytes through activation of phosphoinositide 3-kinase/protein kinase B and AMP-activated protein kinase pathways. Nutr. Res. 2018, 52, 87–97.
- Tolosa, L.; Gomez-Lechon, M.J.; Jimenez, N.; Hervas, D.; Jover, R.; Donato, M.T. Advantageous use of HepaRG cells for the screening and mechanistic study of drug-induced steatosis. Toxicol. Appl. Pharm. 2016, 302, 1–9.
- Teng, Y.; Zhao, Z.; Tasnim, F.; Huang, X.; Yu, H. A scalable and sensitive steatosis chip with long-term perfusion of in situ differentiated HepaRG organoids. Biomaterials 2021, 275, 120904.
- De Gottardi, A.; Vinciguerra, M.; Sgroi, A.; Moukil, M.; Ravier-Dall’Antonia, F.; Pazienza, V.; Pugnale, P.; Foti, M.; Hadengue, A. Microarray analyses and molecular profiling of steatosis induction in immortalized human hepatocytes. Lab. Investig. 2007, 87, 792–806.
- Mahli, A.; Seitz, T.; Beckroge, T.; Freese, K.; Thasler, W.E.; Benkert, M.; Dietrich, P.; Weiskirchen, R.; Bosserhoff, A.; Hellerbrand, C. Bone Morphogenetic Protein-8B Expression is Induced in Steatotic Hepatocytes and Promotes Hepatic Steatosis and Inflammation In Vitro. Cells 2019, 8, 457.
- Prill, S.; Caddeo, A.; Baselli, G.; Jamialahmadi, O.; Dongiovanni, P.; Rametta, R.; Kanebratt, K.P.; Pujia, A.; Pingitore, P.; Mancina, R.M.; et al. The TM6SF2 E167K genetic variant induces lipid biosynthesis and reduces apolipoprotein B secretion in human hepatic 3D spheroids. Sci. Rep. 2019, 9, 11585.
- Parafati, M.; Bae, S.H.; Kirby, R.J.; Fitzek, M.; Iyer, P.; Engkvist, O.; Smith, D.M.; Malany, S. Pluripotent Stem Cell-Derived Hepatocytes Phenotypic Screening Reveals Small Molecules Targeting the CDK2/4-C/EBPalpha/DGAT2 Pathway Preventing ER-Stress Induced Lipid Accumulation. Int. J. Mol. Sci. 2020, 21, 9557.
- Sinton, M.C.; Meseguer-Ripolles, J.; Lucendo-Villarin, B.; Wernig-Zorc, S.; Thomson, J.P.; Carter, R.N.; Lyall, M.J.; Walker, P.D.; Thakker, A.; Meehan, R.R.; et al. A human pluripotent stem cell model for the analysis of metabolic dysfunction in hepatic steatosis. iScience 2021, 24, 101931.
- Aden, D.P.; Fogel, A.; Plotkin, S.; Damjanov, I.; Knowles, B.B. Controlled synthesis of HBsAg in a differentiated human liver carcinoma-derived cell line. Nature 1979, 282, 615–616.
- Mersch-Sundermann, V.; Knasmüller, S.; Wu, X.J.; Darroudi, F.; Kassie, F. Use of a human-derived liver cell line for the detection of cytoprotective, antigenotoxic and cogenotoxic agents. Toxicology 2004, 198, 329–340.
- Moscato, S.; Ronca, F.; Campani, D.; Danti, S. Poly(vinyl alcohol)/gelatin Hydrogels Cultured with HepG2 Cells as a 3D Model of Hepatocellular Carcinoma: A Morphological Study. J. Funct. Biomater. 2015, 6, 16–32.
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007.
- Kugelmas, M.; Hill, D.B.; Vivian, B.; Marsano, L.; McClain, C.J. Cytokines and NASH: A pilot study of the effects of lifestyle modification and vitamin E. Hepatology 2003, 38, 413–419.
- Bahcecioglu, I.H.; Yalniz, M.; Ataseven, H.; Ilhan, N.; Ozercan, I.H.; Seckin, D.; Sahin, K. Levels of serum hyaluronic acid, TNF-alpha and IL-8 in patients with nonalcoholic steatohepatitis. Hepatogastroenterology 2005, 52, 1549–1553.
- Jung, T.W.; Hong, H.C.; Hwang, H.J.; Yoo, H.J.; Baik, S.H.; Choi, K.M. C1q/TNF-Related Protein 9 (CTRP9) attenuates hepatic steatosis via the autophagy-mediated inhibition of endoplasmic reticulum stress. Mol. Cell Endocrinol. 2015, 417, 131–140.
- Peterson, J.M.; Wei, Z.; Seldin, M.M.; Byerly, M.S.; Aja, S.; Wong, G.W. CTRP9 transgenic mice are protected from diet-induced obesity and metabolic dysfunction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R522–R533.
- Mondello, C.; Chiodi, I. Cellular immortalization and neoplastic transformation: Simultaneous, sequential or independent? Telomeres, telomerase or karyotypic variations? Cell Cycle 2013, 12, 1804–1805.
- Vinken, M. Primary hepatocyte cultures for liver disease modeling. Curr. Opin. Toxicol. 2021, 25, 1–5.
- Dooley, S.; Hamzavi, J.; Ciuclan, L.; Godoy, P.; Ilkavets, I.; Ehnert, S.; Ueberham, E.; Gebhardt, R.; Kanzler, S.; Geier, A.; et al. Hepatocyte-specific Smad7 expression attenuates TGF-beta-mediated fibrogenesis and protects against liver damage. Gastroenterology 2008, 135, 642–659.
- Kwon, C.; Sun, J.L.; Jeong, J.H.; Jung, T.W. Humanin attenuates palmitate-induced hepatic lipid accumulation and insulin resistance via AMPK-mediated suppression of the mTOR pathway. Biochem. Biophys. Res. Commun. 2020, 526, 539–545.
- Jung, T.W.; Kang, C.; Goh, J.; Chae, S.I.; Kim, H.C.; Lee, T.J.; Abd El-Aty, A.M.; Jeong, J.H. WISP1 promotes non-alcoholic fatty liver disease and skeletal muscle insulin resistance via TLR4/JNK signaling. J. Cell Physiol. 2018, 233, 6077–6087.
- Jung, T.W.; Kim, H.C.; Abd El-Aty, A.M.; Jeong, J.H. Maresin 1 attenuates NAFLD by suppression of endoplasmic reticulum stress via AMPK-SERCA2b pathway. J. Biol. Chem. 2018, 293, 3981–3988.
- Teschke, R.; Uetrecht, J. Mechanism of idiosyncratic drug induced liver injury (DILI): Unresolved basic issues. Ann. Transl. Med. 2021, 9, 730.
- Huch, M.; Gehart, H.; van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.; Ellis, E.; van Wenum, M.; Fuchs, S.A.; de Ligt, J.; et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 2015, 160, 299–312.
- Broutier, L.; Andersson-Rolf, A.; Hindley, C.J.; Boj, S.F.; Clevers, H.; Koo, B.K.; Huch, M. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nat. Protoc. 2016, 11, 1724–1743.
- Hu, H.; Gehart, H.; Artegiani, B.; LÖpez-Iglesias, C.; Dekkers, F.; Basak, O.; van Es, J.; Chuva de Sousa Lopes, S.M.; Begthel, H.; Korving, J.; et al. Long-Term Expansion of Functional Mouse and Human Hepatocytes as 3D Organoids. Cell 2018, 175, 1591–1606.e19.
- Zhang, K.; Zhang, L.; Liu, W.; Ma, X.; Cen, J.; Sun, Z.; Wang, C.; Feng, S.; Zhang, Z.; Yue, L.; et al. In Vitro Expansion of Primary Human Hepatocytes with Efficient Liver Repopulation Capacity. Cell Stem Cell 2018, 23, 806–819.e4.
- Fu, G.B.; Huang, W.J.; Zeng, M.; Zhou, X.; Wu, H.P.; Liu, C.C.; Wu, H.; Weng, J.; Zhang, H.D.; Cai, Y.C.; et al. Expansion and differentiation of human hepatocyte-derived liver progenitor-like cells and their use for the study of hepatotropic pathogens. Cell Res. 2019, 29, 8–22.
- Vyas, D.; Baptista, P.M.; Brovold, M.; Moran, E.; Gaston, B.; Booth, C.; Samuel, M.; Atala, A.; Soker, S. Self-assembled liver organoids recapitulate hepatobiliary organogenesis in vitro. Hepatology 2018, 67, 750–761.
- Sun, L.; Wang, Y.; Cen, J.; Ma, X.; Cui, L.; Qiu, Z.; Zhang, Z.; Li, H.; Yang, R.Z.; Wang, C.; et al. Modelling liver cancer initiation with organoids derived from directly reprogrammed human hepatocytes. Nat. Cell Biol. 2019, 21, 1015–1026.
- Artegiani, B.; van Voorthuijsen, L.; Lindeboom, R.G.H.; Seinstra, D.; Heo, I.; Tapia, P.; Lopez-Iglesias, C.; Postrach, D.; Dayton, T.; Oka, R.; et al. Probing the Tumor Suppressor Function of BAP1 in CRISPR-Engineered Human Liver Organoids. Cell Stem Cell 2019, 24, 927–943.e6.
- Targher, G.; Mantovani, A.; Byrne, C.D.; Wang, X.B.; Yan, H.D.; Sun, Q.F.; Pan, K.H.; Zheng, K.I.; Chen, Y.P.; Eslam, M.; et al. Risk of severe illness from COVID-19 in patients with metabolic dysfunction-associated fatty liver disease and increased fibrosis scores. Gut 2020, 69, 1545–1547.
- Ji, D.; Qin, E.; Xu, J.; Zhang, D.; Cheng, G.; Wang, Y.; Lau, G. Non-alcoholic fatty liver diseases in patients with COVID-19: A retrospective study. J. Hepatol. 2020, 73, 451–453.
- Tong, X.F.; Wang, Q.Y.; Zhao, X.Y.; Sun, Y.M.; Wu, X.N.; Yang, L.L.; Lu, Z.Z.; Ou, X.J.; Jia, J.D.; You, H. Histological assessment based on liver biopsy: The value and challenges in NASH drug development. Acta Pharm. Sin. 2022, 43, 1200–1209.
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147.
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872.
- Nowak, G.; Ericzon, B.G.; Nava, S.; Jaksch, M.; Westgren, M.; Sumitran-Holgersson, S. Identification of expandable human hepatic progenitors which differentiate into mature hepatic cells in vivo. Gut 2005, 54, 972–979.
- Cai, J.; Zhao, Y.; Liu, Y.; Ye, F.; Song, Z.; Qin, H.; Meng, S.; Chen, Y.; Zhou, R.; Song, X.; et al. Directed differentiation of human embryonic stem cells into functional hepatic cells. Hepatology 2007, 45, 1229–1239.
- Si-Tayeb, K.; Noto, F.K.; Nagaoka, M.; Li, J.; Battle, M.A.; Duris, C.; North, P.E.; Dalton, S.; Duncan, S.A. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 2010, 51, 297–305.
- Huang, P.; He, Z.; Ji, S.; Sun, H.; Xiang, D.; Liu, C.; Hu, Y.; Wang, X.; Hui, L. Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors. Nature 2011, 475, 386–389.
- Sekiya, S.; Suzuki, A. Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature 2011, 475, 390–393.
- Zhu, S.; Rezvani, M.; Harbell, J.; Mattis, A.N.; Wolfe, A.R.; Benet, L.Z.; Willenbring, H.; Ding, S. Mouse liver repopulation with hepatocytes generated from human fibroblasts. Nature 2014, 508, 93–97.
- Ang, L.T.; Tan, A.K.Y.; Autio, M.I.; Goh, S.H.; Choo, S.H.; Lee, K.L.; Tan, J.; Pan, B.; Lee, J.J.H.; Lum, J.J.; et al. A Roadmap for Human Liver Differentiation from Pluripotent Stem Cells. Cell Rep. 2018, 22, 2190–2205.
- Ma, X.; Duan, Y.; Tschudy-Seney, B.; Roll, G.; Behbahan, I.S.; Ahuja, T.P.; Tolstikov, V.; Wang, C.; McGee, J.; Khoobyari, S.; et al. Highly efficient differentiation of functional hepatocytes from human induced pluripotent stem cells. Stem Cells Transl. Med. 2013, 2, 409–419.
- Wu, F.; Wu, D.; Ren, Y.; Huang, Y.; Feng, B.; Zhao, N.; Zhang, T.; Chen, X.; Chen, S.; Xu, A. Generation of hepatobiliary organoids from human induced pluripotent stem cells. J. Hepatol. 2019, 70, 1145–1158.
- Graffmann, N.; Scherer, B.; Adjaye, J. In vitro differentiation of pluripotent stem cells into hepatocyte like cells-Basic principles and current progress. Stem Cell Res. 2022, 61, 102763.
- Hay, D.C.; Zhao, D.; Fletcher, J.; Hewitt, Z.A.; McLean, D.; Urruticoechea-Uriguen, A.; Black, J.R.; Elcombe, C.; Ross, J.A.; Wolf, R.; et al. Efficient differentiation of hepatocytes from human embryonic stem cells exhibiting markers recapitulating liver development in vivo. Stem Cells 2008, 26, 894–902.
- Espejel, S.; Roll, G.R.; McLaughlin, K.J.; Lee, A.Y.; Zhang, J.Y.; Laird, D.J.; Okita, K.; Yamanaka, S.; Willenbring, H. Induced pluripotent stem cell-derived hepatocytes have the functional and proliferative capabilities needed for liver regeneration in mice. J. Clin. Investig. 2010, 120, 3120–3126.
- Touboul, T.; Hannan, N.R.; Corbineau, S.; Martinez, A.; Martinet, C.; Branchereau, S.; Mainot, S.; Strick-Marchand, H.; Pedersen, R.; Di Santo, J.; et al. Generation of functional hepatocytes from human embryonic stem cells under chemically defined conditions that recapitulate liver development. Hepatology 2010, 51, 1754–1765.
- Mun, S.J.; Ryu, J.S.; Lee, M.O.; Son, Y.S.; Oh, S.J.; Cho, H.S.; Son, M.Y.; Kim, D.S.; Kim, S.J.; Yoo, H.J.; et al. Generation of expandable human pluripotent stem cell-derived hepatocyte-like liver organoids. J. Hepatol. 2019, 71, 970–985.
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.-R.; Ueno, Y.; Zheng, Y.-W.; Koike, N.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484.
- Shi, Y.; Inoue, H.; Takahashi, J.; Yamanaka, S. Induced pluripotent stem cell technology: Venturing into the second decade. In Principles of Tissue Engineering; Academic Press: Cambridge, MA, USA, 2020; pp. 435–443.
- Guan, J.; Wang, G.; Wang, J.; Zhang, Z.; Fu, Y.; Cheng, L.; Meng, G.; Lyu, Y.; Zhu, J.; Li, Y.; et al. Chemical reprogramming of human somatic cells to pluripotent stem cells. Nature 2022, 605, 325–331.
This entry is offline, you can click here to edit this entry!