Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Apoptosis is one of the most well-studied and characterized programmed cell death mechanisms. The detailed characterization of molecular interactions involved in apoptosis, and the growing amount of related quantitative data, has encouraged computational and systems biologists to develop mathematical models of apoptosis.
- cell death
- apoptosis
- mathematical models
- computational analysis
1. Introduction
Mathematical modeling is a powerful tool that allows one to connect molecular biology to cell physiology by associating the qualitative and quantitative features of dynamical molecular networks with signal–response curves measured by cell biologists [1]. Mathematical and systems-oriented approaches have been successfully applied to describe the dynamics of complex molecular networks that control cell cycle [2][3], nutrient signaling [4], checkpoints [5], signaling dysregulation in cancer [6], and cell death [7][8][9][10][11][12][13]. Systems-oriented mathematical approaches are especially useful for analyzing complex systems that cannot be understood by intuitive reasoning. Undoubtedly, cell death regulation is one such molecular mechanism that cannot be fully understood without mathematical modeling.
Cell death mechanisms are directly involved in regulations of tissue homeostasis, inflammation, immunity, development and other physiological processes [14]. Characterization of new genes and molecular components, involved in signaling pathways by regulating cell death, continues to progress. A detailed characterization of cell death regulation can help identify novel targets and develop effective therapeutic protocols to strike acquired drug resistance in cancer cells. Accurate predictive mechanistic models of complex molecular networks regulating cell death can be used to test the effects of new drugs on the system, and to search for synergistic drug combinations and effective treatment protocols. Different modeling approaches have been already successfully applied to model extensive cell death molecular networks. Ordinary differential equations (ODEs), Boolean logic, pharmacokinetic-pharmacodynamic (PK-PD), Petri nets, agent-based modeling (ABM), cellular automata and hybrid approaches are the common choices available to model molecular mechanisms involved in cell death control, decisions and execution [6][12][13][15][16][17][18][19].
2. Mathematical Models of Apoptosis
Apoptosis is one of the most well-studied and characterized programmed cell death mechanisms. The detailed characterization of molecular interactions involved in apoptosis, and the growing amount of related quantitative data, has encouraged computational and systems biologists to develop mathematical models of apoptosis [12][13][17]. Over the last twenty years, several dozen mathematical models of apoptosis regulation have been described. These apoptosis models aim to explain different data or effects of different treatments on cell death. While the core molecular components regulating apoptosis are shared by all models, variations in molecular circuit designs, components, data, mathematical approaches, and study goals make each model a unique tool to study apoptosis. Most often, molecular mechanisms of apoptosis are mathematically represented using ODEs [7][20][21][22][23][24][25][26][27][28][29][30], Boolean logics [31][32][33], and Petri nets [16]; other computational approaches have also been applied [18][34][35].
The execution core of apoptosis regulation involves a family of proteases termed caspases. Caspases can be separated into the following two groups: effector or executioner caspases (caspase-3, -6, -7), and active initiator caspases (e.g., caspases-8, -9). Activation of the caspases initiates the cleavage of several important cellular proteins, such as actin and nuclear lamins, which results in cell body and nuclear shrinkage and cell death [36]. Apoptosis can be processed through mitochondria-dependent (intrinsic apoptosis) and mitochondria-independent (extrinsic apoptosis) caspase-3 activation pathways [14]. The core components involved in these two pathways are commonly included in all mathematical models of apoptosis and can be found in the earliest mathematical models of apoptosis [7].
Extrinsic apoptosis is characterized by high amounts of active caspase-8 that activates the downstream effectors caspase-3, caspase-6, and caspase-7. The activation of caspase-8 is receptor-mediated, which occurs upon receipt of a death signal that is processed by a surface death receptor such as FAS (a member of the tumor necrosis factor gene superfamily) [14]. Therefore, extrinsic apoptosis is a receptor-mediated cell death mechanism, as shown in Figure 1 (left). By contrast, intrinsic apoptosis can be executed even in cells with lower levels of active caspase-8 but requires an additional amplification that involves activation of the pro-apoptotic functions of the mitochondria. For example, stress-related factors (e.g., DNA damage) can induce activation of the executioner caspases via a mitochondria-dependent pathway in the absence of an external death signal [37] (Figure 1, right panel). The mitochondria-dependent pathway begins with the cleavage of anti-apoptotic Bcl-2 family members, which causes the aggregation of pro-apoptotic proteins such as Bax and Bak. Aggregation of pro-apoptotic proteins is followed by the release of cytochrome c from the mitochondria, which induces the formation of a large protein complex known as the apoptosome. The apoptosome recruits and activates caspase-9, allowing it to cleave the downstream effectors pro-caspase-3, pro-caspase-6, and pro-caspase-7. Notably, the expression of anti-apoptotic Bcl-2 family members can block the intrinsic apoptosis signaling in cells. By contrast, extrinsic apoptosis cannot be blocked by the expression of high levels of Bcl-2 proteins because large amounts of caspase-8 are already generated.
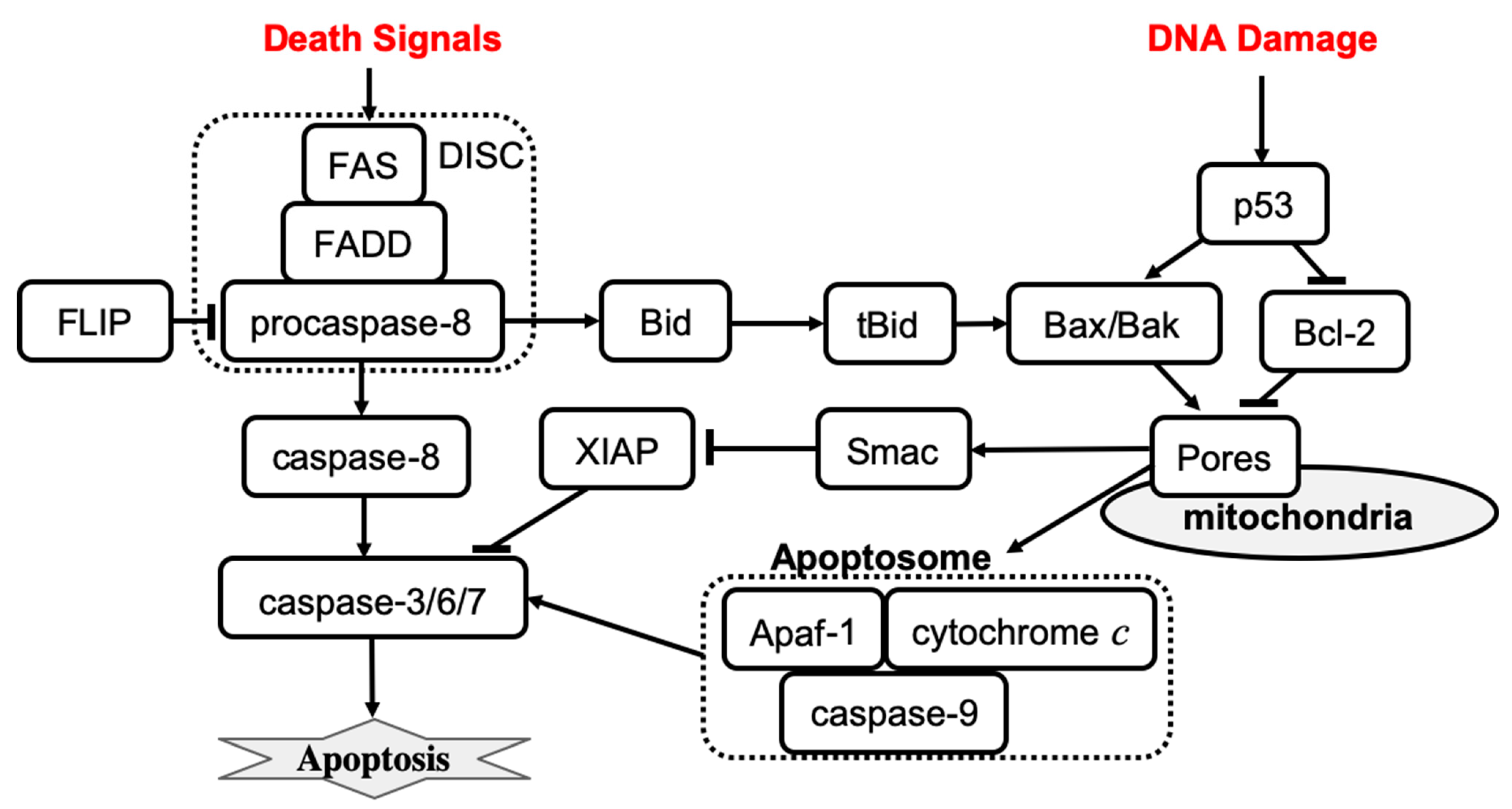
Figure 1. The mechanisms of receptor-induced apoptosis (left) and stress-mediated, mitochondria-dependent apoptosis (right). Solid lines represent activation (arrowhead) and inhibition (bar head) influences.
The earliest mathematical models of apoptosis described both mitochondria-dependent and independent death activation pathways. In early 2000, Fussenegger et al. published a mechanistic ODE-based mathematical model of apoptosis that describes both receptor-mediated and stress-induced caspase activation mechanisms [7]. The receptor-mediated feature of the model describes the FAS surface receptor that activates procaspase-8. Activation of apoptosis initiator caspases involves the following reactions: the binding of an extracellular death ligand to the FAS receptor, the binding of FAS-associated death domain (FADD) protein to the FAS death domain, and the binding of caspase-8 to a domain on FADD that enables caspase-8 activation by proteolytic cleavage. Each binding process is described by a specific rate parameter in the model. Simulation results show that about 50% of procaspase-8 is activated within two hours after the death signal is received. After procaspase-8 activation, the executioner caspase is activated within minutes, and then the initiation of procaspase-9 occurs with the lag time ~20–30 min. The activation curves have a sigmoidal shape indicating, that the transition between the inactive to the active state is characterized by a threshold. If the binding between FADD and clustered FAS death domains is disrupted, then only <0.1% of active caspase-8 is observed upon receipt of the death signal, which is consistent with experimental observations [38].
Fussenegger’s model of stress-mediated apoptosis regulation describes the activation of procaspase-9 by cytosolic cytochrome c, and the apoptotic protease-activating factor 1 (Apaf-1) complex. Activated caspase-9 then activates apoptosis executioner caspases at some specific rate. Formation of the Apaf-1–cytochrome c complex is inhibited by antiapoptotic Bcl-2 family members such as Bcl-xL. Proapoptotic Bcl-2 family members (e.g., Bax, Bak) can bind to antiapoptotic family members and remove their inhibitory effect. The ratio of anti- versus pro-apoptotic Bcl-2 family members is controlled by the p53 transcription factor that is activated in cells under stress conditions. Simulation results of stress-induced caspase activation dynamics were consistent with experimental observations [39]. Specifically, the model shows that cytochrome c is released within 10 min after a stress death signal is received, which results in procaspase-9 activation, 35–40% of the executioner caspase being active within 1 h, and 70% of the executioner caspase being active at 2 h. In addition, simulations revealed that the active fraction of both initiator and executioner caspases is reduced in p53 mutant cells as compared to wild-type cells. Overexpression of antiapoptotic Bcl-2 family members is predicted to block the activation of procaspase-9. The model also confirms that the ratio of anti- versus pro-apoptotic Bcl-2 family members determines whether or not executioner caspases will be activated. The model was then used to predict the effects of combined therapies based on simultaneous receptor- and stress-induced caspase activation.
The model developed by Fussenegger et al. was successful in explaining qualitative experimental observations. However, more quantitative data would be required to complete the model calibration. Quantitative information on reaction rates and molecular concentrations is required to perform reliable mathematical simulations of signal transduction in the apoptosis regulatory network. In 2004, Eissing et al. developed a reduced receptor-induced apoptosis, using parameter values from the literature to evaluate the system behavior within a wide range of parameters [20]. The model revealed that caspase activity remains low for a time that is inversely proportional to the stimulus strength, followed by a steep rise in activity when the input exceeds the threshold; caspase activity then ceases at some maximum level. Bifurcation analysis of the model confirmed that the apoptosis regulation system exhibits a bistable behavior. The same year, Bentele et al. developed a data-based model of receptor-induced apoptosis with parameters estimated on the basis of quantitative experimental data [22]. The time series data for concentrations of 15 different molecules after activation of FAS receptors were used to calibrate the core model of the FAS-induced apoptosis. In addition, data from distinct apoptosis activation scenarios in response to different initial values of ligand concentration were used to improve the estimation of model parameters. The model predicted that apoptosis is not executed when a ligand–receptor concentration ratio is below a critical value, which was also confirmed by experimental observations. In conclusion, Bentele et al. proposed a threshold mechanism for induction of receptor-induced apoptosis. A year later, Hua et al. published a FAS-induced apoptosis model to investigate the effects of altering the level of Bcl-2 on the kinetics of caspase-3 activation [40]. The model predicts that Bcl-2 blocks the mitochondrial pathway by binding to proapoptotic Bax, Bak, and tBid proteins. Further, the model predicts that apoptosis signaling flow can be switched between mitochondria-dependent and mitochondria-independent pathways by varying molecular component levels without changing network structure.
In 2006, Legewie et al. developed a quantitative kinetic model of intrinsic (stress-induced) apoptosis, which displays an all-or-none behavior of caspase activation in response to an apoptotic stimulus [21]. The model helped to identify the positive feedback mechanism that allows cells to achieve ultrasensitivity and bistability in cell death decision making. The pathway molecular regulators that control the apoptotic threshold stimulus and integrate multiple inputs into an all-or-none caspase output were also determined. Time-course simulation results agreed with experimental observations that the induction of maximal caspase-3 cleavage after exogenous addition of cytochrome c occurs within ~15–60 min. Furthermore, cytochrome c-induced activation of caspase-3 was observed to be bistable and irreversible. The bistable and irreversible caspase-3 activation arises in the system due to XIAP-mediated feedback that cooperates with caspase-9 cleavage by caspase-3. X-linked inhibitor of apoptosis (XIAP) inhibits the catalytic activities of caspase-9 and caspase-3 through reversible binding. The feedback cleavage of caspase-9 by caspase-3 leads to autoamplification of the apoptotic signal. Simulation results show that XIAP-mediated feedback is observed only if caspase-9 and caspase-3 compete for binding to XIAP. Depletion and re-addition experiments using different Apaf-1, caspase-3, caspase-9, and/or XIAP concentrations were proposed to test the all-or-none caspase activation.
Also in 2006, Rehm et al. published a computational model of apoptosome-dependent caspase activation based on biochemical data from HeLa cells [23]. The model predicts that the all-or-none apoptotic response depends on caspase-3-dependent feedback signaling and XIAP, which was then verified quantitatively using single-cell experiments with a caspase fluorescence resonance energy transfer substrate. A concentration threshold of XIAP between 0.15 and 0.30 μM, controlling the substrate cleavage by effector caspases, was identified. The model suggested that high levels of XIAP may promote apoptosis resistance and sublethal caspase activation. This result agrees with a computational analysis that was performed earlier, which also suggested that the inhibitor of apoptosis plays an important role in both the induction and prevention of apoptosis [41]. Conversely, Bagci et al. proposed a mathematical model of mitochondria-dependent apoptosis to study both the role of Bax and Bcl-2 synthesis, degradation rates and the number of mitochondrial permeability transition pores involved in the cell response to a death signal [42]. The main finding was that the transition from bistable to monostable (survival) cell behavior is controlled by the synthesis and degradation rates of Bax and Bcl-2 and by the number of mitochondrial permeability transition pores. Also, the model results suggested that cooperative apoptosome formation is a much more robust mechanism to induce bistability than feedback mechanisms involving, for example, the inhibition of caspase-3 by the inhibitor of apoptosis. Later, Chen and Cui et al. analyzed the robustness of Bax and Bcl-2 apoptotic switches using both deterministic and stochastic models [35][43][44]. These mechanisms were confirmed to be bistable and robust to noise and wide ranges of parameter variation.
Albeck et al. developed a mathematical model of extrinsic, receptor-induced apoptosis to explain the molecular mechanism of the variable-delay, snap-action switch function that determines the cell choice between life and death [24]. The model was calibrated by experimental data collected from live-cell imaging, flow cytometry, and immunoblotting of cells perturbed by protein depletion and overexpression. The model was then used to reveal the mechanism by which a steady and gradual increase in caspase-8 activity is converted into a snap-action downstream signal. Permeabilization of the mitochondrial membrane and relocalization of proteins are the key factors in the extrinsic apoptosis network by which a graded signal that activates caspase-8 and promotes the formation of pores in the mitochondrial membrane is transformed into an all-or-none death decision. Importantly, such snap-action behavior at the level of the mitochondrial outer membrane permeabilization occurs independently of caspase-dependent feedback mechanisms. The formation of pores in the mitochondrial membrane involves the pore-forming proteins Bax and Bak that can self-assemble into transmembrane pores, which are antagonized by anti-apoptotic Bcl-2 proteins [45]. Cytochrome c is released into the cytosol when the level of active pore-forming proteins exceeds the threshold set by anti-apoptotic Bcl-2 proteins. Using experimental and modeling techniques, Spencer et al. demonstrated that cell-to-cell variability in time-to-death significantly depends on the activation rate of the tBid protein that activates the pore-forming proteins, Bax and Bak [30]. Therefore, in the case of receptor-mediated apoptosis, the timing and probability of death relies on the differences in the protein levels that can be caused, for example, by noise in gene expression. Furthermore, the stochastic protein turnover in a receptor-mediated apoptosis model can result in fractional killing [46].
Later models were developed to investigate crosstalk between apoptosis regulation and NF-κB pathways [29], the estrogen signaling network [28], endoplasmic pathways [25], and autophagy regulation [26]. Neumann et al. described a model of the crosstalk between receptor-mediated apoptosis regulation and NF-κB signaling that are activated by the same receptor in parallel to the apoptotic signaling and on a similar time scale [29]. Model and experimental analysis suggested that the balance between apoptotic and NF-κB signaling is shaped by the proteins that regulate the assembly dynamics of the death-inducing signaling complex (DISC). Therefore, the assembly of DISC acts as a signal processor, determining life/death decisions in a nonlinear manner. Tyson et al. provided a roadmap for a detailed mathematical model that would allow researchers to characterize the crosstalk among the estrogen signaling network, apoptosis, autophagy, and cell cycle regulations in breast epithelial cells [28]. Later, the same research lab published a detailed mathematical model to examine the decision process that moves a cell from autophagy to apoptosis [26]. The model was successful in explaining quantitative time-course data of autophagy and apoptosis under cisplatin treatment. Further, the model allows for characterization of the prosurvival and prodeath cell responses to cytotoxic stress. Also, in 2012, Hong et al. published a model of cisplatin-induced apoptosis that integrates the death receptor pathway, and mitochondrial and endoplasmic reticulum stress response mechanisms [25]. The model predicts the relative contribution of each signaling pathway to apoptosis. Simulation results revealed that the mitochondrial and death receptor pathways as well as crosstalk among pathways make the greatest contribution to the level of apoptosis, whereas the contribution of the endoplasmic reticulum stress pathway is negligible.
The Role of p53 in Apoptosis
The tumor suppressor gene p53 (TP53) has been reported as an upregulated modulator of apoptosis and as a driver of cell fate transition from cell cycle arrest to apoptosis [47]. Mathematical models that characterize the p53 contribution to apoptosis have been developed by several groups [7][25][27][42][48]. p53 targets many genes regulating cell apoptosis, including BCL2 and BAX genes [49]. Computational study of apoptosis regulation shows that the balance between anti- and proapoptotic Bcl-2 family members is altered in p53 mutant cells [7]. Also, the active fraction of both initiator and executioner caspases is reduced in p53 mutant cells as compared with wild-type cells. The mathematical model also predicts that overexpression of the death ligand and the FAS receptor can be used to initiate executioner caspase activation in p53 mutant cells [7]. Bagci et al. have shown that apoptosis is not sensitive to caspase-3 activation when p53 expression is low, and that bistability to apoptotic stimuli is observed when p53 level is high [42]. Predictions from this apoptosis model agree with experimental data [50]. Another study reported that inhibition of p53 protects against cisplatin-induced apoptosis [25]. Cisplatin induces DNA damage that results in the phosphorylation and activation of p53. There, the activation of Bax by p53 induces mitochondrial membrane permeabilization and apoptosis [51]. Also, p53 mediates caspase-2 activation and the mitochondrial release of apoptosis-inducing factor. The model predicts time courses for p53, caspase-2, Bax activation, apoptosis-inducing factor release and apoptosis activation. Simulation results agree with experimental data that p53 inhibition prevents the mitochondrial release of apoptosis-inducing factor and cisplatin-induced apoptosis [52]. Overexpression of p53 results in caspase-2 activation and also the mitochondrial release of apoptosis-inducing factors [52].
Ballweg et al. developed a mathematical model that integrates p53 signaling, cisplatin-induced events, and apoptosis regulation that was used to study the dynamics of fractional killing induced by cytotoxic drugs [27]. Many drugs activate not only apoptosis execution signaling but also expression of anti-apoptotic genes, which results only in fractional killing amongst a population of treated cells [53]. Thus, fractional killing may occur due to crosstalk between the apoptosis and survival pathways [54]. The model predicts that the probability of apoptosis depends on the dynamics of p53 and the rate of p53 activation determines the cell fate [27]. Slow activation of p53 results in cell survival, whereas fast p53 activation induces cell death. This result also agrees with the experimental observation showing that apoptotic cells accumulate p53 much earlier than cells that survive the treatment [53]. In the model, activation of Bax and subsequent execution of apoptosis occur when the level of p53 exceeds a threshold value. However, the apoptosis initiation threshold depends on the inhibitor of apoptosis, cIAP. Cells with an elevated level of cIAP require a higher level of p53 to induce apoptosis. Because the level of apoptosis regulator cIAP increases with time, the rate of p53 activation plays an important role in the determination of cell fate. Cell-to-cell variability due to stochastic gene expression and environmental noise can also set different apoptosis initiation thresholds in different cells, resulting in fractional killing.
Up to this point, mathematical models of apoptosis that use ODEs to describe the mechanism of cell death (apoptosis) regulation have been reviewed. However, other mathematical approaches have been also used to study apoptosis regulation [16][18][31][32][33][34]. Several apoptosis models have been developed using a Boolean (logical) approach that can analyze extensive regulatory networks with many molecular components and their interactions [31][32][33]. Schlatter et al. developed an apoptosis regulation model that comprises 86 nodes and 125 interactions [31]. Mai et al. developed a model that describes 37 internal states of signaling molecules involved in apoptosis regulation, 2 extracellular signal inputs, and the DNA damage event as an output [32]. Calzone et al. developed a model to study crosstalk between receptor-mediated apoptosis regulation, NFκB pro-survival pathways, and RIP1-dependent necroptosis regulation [33]. These models were used to characterize feedback loops in the apoptosis regulation network structure.
While Boolean models are excellent tools to reproduce the qualitative behavior of a regulatory network, they are weak at addressing detailed quantitative questions about molecular mechanisms [19]. Petri nets have been applied to analyze and validate a qualitative model of extensive apoptosis regulation [16]. Agent-based modeling turned out to be a more appropriate approach for modeling the death-inducing signaling complex assembly than an ODE-based model that must describe a large number of intermediate products involved in DISC assembly [34]. A cellular automata approach has been applied to study apoptosis blocking in the immunological response of T cells by varying the inhibitor actions such as FLIP and IAP [18]. The model predicts that only joint suppression of both FLIP and IAP apoptosis inhibitors can effectively act to kill cancer cells through apoptosis.
In conclusion, comprehensive data and extensive experimental characterization of apoptosis allowed computational and systems biologists to develop several mathematical models of apoptosis regulation. These models not only increase our understanding of mechanisms of apoptosis execution induced by stress or signals, but also predict perturbations that can prevent or enhance apoptosis. An accurate mathematical model of apoptosis can help find novel combinations of existing therapies that can induce the death of cancer cells using low doses. Further studies that integrate apoptosis with other cell death regulations will help to understand the cell death decision mechanism that determines the execution of a specific cell death fate.
This entry is adapted from the peer-reviewed paper 10.3390/e24101402
References
- Tyson, J.J.; Novak, B. A Dynamical Paradigm for Molecular Cell Biology. Trends. Cell Biol. 2020, 30, 504–515.
- Kraikivski, P.; Chen, K.C.; Laomettachit, T.; Murali, T.; Tyson, J.J. From START to FINISH: Computational analysis of cell cycle control in budding yeast. NPJ Syst. Biol. Appl. 2015, 1, 15016.
- Shafiekhani, S.; Kraikivski, P.; Gheibi, N.; Ahmadian, M.; Jafari, A. Dynamical analysis of the fission yeast cell cycle via Markov chain. Curr. Genet. 2021, 67, 785–797.
- Jalihal, A.P.; Kraikivski, P.; Murali, T.; Tyson, J.J. Modeling and Analysis of the Macronutrient Signaling Network in Budding Yeast. Mol. Biol. Cell 2021, 32, ar20.
- Jung, Y.; Kraikivski, P.; Shafiekhani, S.; Terhune, S.S.; Dash, R.K. Crosstalk between Plk1, p53, cell cycle, and G2/M DNA damage checkpoint regulation in cancer: Computational modeling and analysis. NPJ Syst. Biol. Appl. 2021, 7, 46.
- Clarke, R.; Kraikivski, P.; Jones, B.C.; Sevigny, C.M.; Sengupta, S.; Wang, Y. A systems biology approach to discovering pathway signaling dysregulation in metastasis. Cancer Metastasis Rev. 2020, 39, 903–918.
- Fussenegger, M.; Bailey, J.E.; Varner, J. A mathematical model of caspase function in apoptosis. Nat. Biotechnol. 2000, 18, 768–774.
- Xu, F.; Yin, Z.; Zhu, L.; Jin, J.; He, Q.; Li, X.; Shuai, J. Oscillations Governed by the Incoherent Dynamics in Necroptotic Signaling. Front. Phys. 2021, 9, 482.
- Zhu, L.; Li, X.; Xu, F.; Yin, Z.; Jin, J.; Liu, Z.; Qi, H.; Shuai, J. Network modeling-based identification of the switching targets between pyroptosis and secondary pyroptosis. Chaos Solitons Fractals 2022, 155, 111724.
- Konstorum, A.; Tesfay, L.; Paul, B.T.; Torti, F.M.; Laubenbacher, R.C.; Torti, S.V. Systems biology of ferroptosis: A modeling approach. J. Theor. Biol. 2020, 493, 110222.
- Checcoli, A.; Pol, J.G.; Naldi, A.; Noël, V.; Barillot, E.; Kroemer, G.; Thieffry, D.; Calzone, L.; Stoll, G. Dynamical Boolean Modeling of Immunogenic Cell Death. Front. Physiol. 2020, 11, 590479.
- Lavrik, I.N. Systems biology of apoptosis signaling networks. Curr. Opin. Biotechnol. 2010, 21, 551–555.
- Lavrik, I.N.; Eils, R.; Fricker, N.; Pforr, C.; Krammer, P.H. Understanding apoptosis by systems biology approaches. Mol. Biosyst. 2009, 5, 1105–1111.
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337.
- Aldridge, B.B.; Burke, J.M.; Lauffenburger, D.A.; Sorger, P.K. Physicochemical modelling of cell signalling pathways. Nat. Cell Biol. 2006, 8, 1195–1203.
- Heiner, M.; Koch, I.; Will, J. Model validation of biological pathways using Petri nets—Demonstrated for apoptosis. Biosystems 2004, 75, 15–28.
- Schleich, K.; Lavrik, I.N. Mathematical modeling of apoptosis. Cell Commun. Signal. 2013, 11, 44.
- Apte, A.; Bonchev, D.; Fong, S. Cellular automata modeling of FASL-initiated apoptosis. Chem. Biodivers. 2010, 7, 1163–1172.
- Tyson, J.J.; Laomettachit, T.; Kraikivski, P. Modeling the dynamic behavior of biochemical regulatory networks. J. Theor. Biol. 2019, 462, 514–527.
- Eissing, T.; Conzelmann, H.; Gilles, E.D.; Allgower, F.; Bullinger, E.; Scheurich, P. Bistability analyses of a caspase activation model for receptor-induced apoptosis. J. Biol. Chem. 2004, 279, 36892–36897.
- Legewie, S.; Bluthgen, N.; Herzel, H. Mathematical modeling identifies inhibitors of apoptosis as mediators of positive feedback and bistability. PLoS Comput. Biol. 2006, 2, e120.
- Bentele, M.; Lavrik, I.; Ulrich, M.; Stosser, S.; Heermann, D.; Kalthoff, H.; Krammer, P.; Eils, R. Mathematical modeling reveals threshold mechanism in CD95-induced apoptosis. J. Cell Biol. 2004, 166, 839–851.
- Rehm, M.; Huber, H.J.; Dussmann, H.; Prehn, J.H. Systems analysis of effector caspase activation and its control by X-linked inhibitor of apoptosis protein. EMBO J. 2006, 25, 4338–4349.
- Albeck, J.G.; Burke, J.M.; Spencer, S.L.; Lauffenburger, D.A.; Sorger, P.K. Modeling a snap-action, variable-delay switch controlling extrinsic cell death. PLoS Biol. 2008, 6, 2831–2852.
- Hong, J.-Y.; Kim, G.-H.; Kim, J.-W.; Kwon, S.-S.; Sato, E.F.; Cho, K.-H.; Shim, E.B. Computational modeling of apoptotic signaling pathways induced by cisplatin. BMC Syst. Biol. 2012, 6, 122.
- Tavassoly, I.; Parmar, J.; Shajahan-Haq, A.; Clarke, R.; Baumann, W.T.; Tyson, J.J. Dynamic Modeling of the Interaction Between Autophagy and Apoptosis in Mammalian Cells. CPT Pharmacomet. Syst. Pharmacol. 2015, 4, 263–272.
- Ballweg, R.; Paek, A.L.; Zhang, T. A dynamical framework for complex fractional killing. Sci. Rep. 2017, 7, 8002.
- Tyson, J.J.; Baumann, W.T.; Chen, C.; Verdugo, A.; Tavassoly, I.; Wang, Y.; Weiner, L.M.; Clarke, R. Dynamic modelling of oestrogen signalling and cell fate in breast cancer cells. Nat. Rev. Cancer 2011, 11, 523–532.
- Neumann, L.; Pforr, C.; Beaudouin, J.; Pappa, A.; Fricker, N.; Krammer, P.H.; Lavrik, I.N.; Eils, R. Dynamics within the CD95 death-inducing signaling complex decide life and death of cells. Mol. Syst. Biol. 2010, 6, 352.
- Spencer, S.L.; Gaudet, S.; Albeck, J.G.; Burke, J.M.; Sorger, P.K. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 2009, 459, 428–432.
- Schlatter, R.; Schmich, K.; Avalos Vizcarra, I.; Scheurich, P.; Sauter, T.; Borner, C.; Ederer, M.; Merfort, I.; Sawodny, O. ON/OFF and beyond—A boolean model of apoptosis. PLoS Comput. Biol. 2009, 5, e1000595.
- Mai, Z.; Liu, H. Boolean network-based analysis of the apoptosis network: Irreversible apoptosis and stable surviving. J. Theor. Biol. 2009, 259, 760–769.
- Calzone, L.; Tournier, L.; Fourquet, S.; Thieffry, D.; Zhivotovsky, B.; Barillot, E.; Zinovyev, A. Mathematical modelling of cell-fate decision in response to death receptor engagement. PLoS. Comput. Biol. 2010, 6, e1000702.
- Schleich, K.; Warnken, U.; Fricker, N.; Öztürk, S.; Richter, P.; Kammerer, K.; Schnölzer, M.; Krammer, P.H.; Lavrik, I.N. Stoichiometry of the CD95 death-inducing signaling complex: Experimental and modeling evidence for a death effector domain chain model. Mol. Cell 2012, 47, 306–319.
- Chen, C.; Cui, J.; Lu, H.; Wang, R.; Zhang, S.; Shen, P. Modeling of the role of a Bax-activation switch in the mitochondrial apoptosis decision. Biophys. J. 2007, 92, 4304–4315.
- Krammer, P.H.; Arnold, R.; Lavrik, I.N. Life and death in peripheral T cells. Nat. Rev. Immunol. 2007, 7, 532–542.
- Norbury, J.C.; Zhivotovsky, B. DNA damage-induced apoptosis. Oncogene 2004, 23, 2797–2808.
- Chinnaiyan, A.M.; O’Rourke, K.; Tewari, M.; Dixit, V.M. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995, 81, 505–512.
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, D.D. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science 1997, 275, 1132–1136.
- Hua, F.; Cornejo, M.G.; Cardone, M.H.; Stokes, C.L.; Lauffenburger, D.A. Effects of Bcl-2 levels on Fas signaling-induced caspase-3 activation: Molecular genetic tests of computational model predictions. J. Immunol. 2005, 175, 985–995.
- Stucki, W.J.; Simon, H.U. Mathematical modeling of the regulation of caspase-3 activation and degradation. J. Theor. Biol. 2005, 234, 123–131.
- Bagci, E.; Vodovotz, Y.; Billiar, T.; Ermentrout, G.; Bahar, I. Bistability in apoptosis: Roles of bax, bcl-2, and mitochondrial permeability transition pores. Biophys. J. 2006, 90, 1546–1559.
- Chen, C.; Cui, J.; Zhang, W.; Shen, P. Robustness analysis identifies the plausible model of the Bcl-2 apoptotic switch. FEBS Lett. 2007, 581, 5143–5150.
- Cui, J.; Chen, C.; Lu, H.; Sun, T.; Shen, P. Two independent positive feedbacks and bistability in the Bcl-2 apoptotic switch. PLoS ONE 2008, 3, e1469.
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59.
- Bertaux, F.; Stoma, S.; Drasdo, D.; Batt, G. Modeling dynamics of cell-to-cell variability in TRAIL-induced apoptosis explains fractional killing and predicts reversible resistance. PLoS Comput. Biol. 2014, 10, e1003893.
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956.
- Kim, E.; Kim, J.Y.; Lee, J.Y. Mathematical Modeling of p53 Pathways. Int. J. Mol. Sci. 2019, 20, 5179.
- Miyashita, T.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299.
- Katiyar, S.K.; Roy, A.M.; Baliga, M.S. Silymarin induces apoptosis primarily through a p53-dependent pathway involving Bcl-2/Bax, cytochrome c release, and caspase activation. Mol. Cancer Ther. 2005, 4, 207–216.
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014.
- Seth, R.; Yang, C.; Kaushal, V.; Shah, S.V.; Kaushal, G.P. p53-dependent caspase-2 activation in mitochondrial release of apoptosis-inducing factor and its role in renal tubular epithelial cell injury. J. Biol. Chem. 2005, 280, 31230–31239.
- Paek, A.L.; Liu, J.C.; Loewer, A.; Forrester, W.C.; Lahav, G. Cell-to-Cell Variation in p53 Dynamics Leads to Fractional Killing. Cell 2016, 165, 631–642.
- Falschlehner, C.; Emmerich, C.H.; Gerlach, B.; Walczak, H. TRAIL signalling: Decisions between life and death. Int. J. Biochem. Cell. Biol. 2007, 39, 1462–1475.
This entry is offline, you can click here to edit this entry!