Dosage compensation is the process by which organisms equalize the expression of genes between members of different biological sexes. Across species, different sexes are often characterized by different types and numbers of sex chromosomes. In order to neutralize the large difference in gene dosage produced by differing numbers of sex chromosomes among the sexes, various evolutionary branches have acquired various methods to equalize gene expression among the sexes. Because sex chromosomes contain different numbers of genes, different species of organisms have developed different mechanisms to cope with this inequality. Replicating the actual gene is impossible; thus organisms instead equalize the expression from each gene. For example, in humans, females (XX) silence the transcription of one X chromosome of each pair, and transcribe all information from the other, expressed X chromosome. Thus, human females have the same number of expressed X-linked genes as do human males (XY), both sexes having essentially one X chromosome per cell, from which to transcribe and express genes. There are three main mechanisms of achieving dosage compensation which are widely documented in the literature and which are common to most species. These include random inactivation of one female X chromosome (as observed in Mus musculus; this is called X-inactivation), a two-fold increase in the transcription of a single male X chromosome (as observed in Drosophila melanogaster), and decreased transcription by half in both of the X chromosomes of a hermaphroditic organism (as observed in Caenorhabditis elegans). These mechanisms have been widely studied and manipulated in model organisms commonly used in the laboratory research setting. A summary of these forms of dosage compensation is illustrated below. However, there are also other less common forms of dosage compensation, which are not as widely researched and are sometimes specific to only one species (as observed in certain bird and monotreme species).
- model organisms
- melanogaster
- musculus
1. Random Inactivation of One ♀ X
One logical way to equalize gene expression amongst males and females that follow a XX/XY sex differentiation scheme would be to decrease or altogether eliminate the expression of one of the X chromosomes in an XX, or female, homogametic individual, such that both males and females then express only one X chromosome. This is the case in many mammalian organisms, including humans and mice.[1]
The evidence for this mechanism of dosage compensation was discovered prior to scientists’ understanding of what its implications were. In 1949, Murray Barr and Ewert Bertram published data describing the presence of “nucleolar satellites,[2] which they observed were present in the mature somatic tissue of different female species. Further characterization of these satellites revealed that they were actually packages of condensed heterochromatin, but a decade would pass before scientists grasped the significance of this specialized DNA.
Then, in 1959 Susumu Ohno proved that these satellite-like structures found exclusively in female cells were actually derived from female X chromosomes.[3] He called these structures Barr bodies after one of the investigators who originally documented their existence. Ohno’s studies of Barr bodies in female mammals with multiple X chromosomes revealed that such females used Barr bodies to inactivate all but one of their X chromosomes. Thus, Ohno described the “n-1” rule to predict the number of Barr bodies in a female with n number of X chromosomes in her karyotype.[3]
Simultaneously, Mary F. Lyon began investigating manipulations of X-linked traits that had phenotypically visible consequences, particularly in mice, whose fur color is a trait intimately linked to the X chromosome. Building on work done by Ohno and his colleagues, Lyon eventually proved that either the maternal or paternal X chromosome is randomly inactivated in every cell of the female body in the species she was studying,[4] which explained the heterogeneous fur patterns she observed in her mosaic mice. This process is known as X-inactivation, and is sometimes referred to as “lyonization”.[1] This discovery can be easily extrapolated to explain the mixed color patterns observed in the coats of tortoiseshell cats. The fur patterns characteristic of tortoiseshell cats are found almost exclusively in females, because only they randomly inactivate one X chromosome in every somatic hair cell.[5] Thus, presuming that hair color determining genes are X-linked, it makes sense that whether the maternal or paternal X chromosome is inactivated in a particular hair cell can result in differential fur color expression.
Compounding on Lyon's discoveries, in 1962 Ernest Beutler used female fibroblast cell lineages grown in culture to demonstrate the heritability of lyonization or random X-inactivation.[6] By analyzing the differential expression of two existing, viable alleles for the X-linked enzyme glucose-6-phosphate dehydrogenase (G6PD) gene, Beutler observed that the inactivation of the gene was heritable across passaged generations of the cells.[7]
This pattern of dosage compensation, caused by random X-inactivation, is regulated across development in female mammals, following concerted patterns throughout development; for example, at the beginning of most female mammal development, both X chromosomes are initially expressed, but gradually undergo epigenetic processes to eventually achieve random inactivation of one X.[7] In germ cells, inactivated X chromosomes are then once again activated to ensure their expression in gametes produced by female mammals.[1]
Thus, dosage compensation in mammals is largely achieved through the silencing of one of two female X chromosomes via X-inactivation. This process involves histone tail modifications, DNA methylation patterns, and reorganization of large-scale chromatin structure encoded by the X-ist gene.[1] In spite of these extensive modifications, not all genes along the X chromosome are subject to X-inactivation; active expression at some loci is required for homologous recombination with the pseudo-autosomal region (PAR) of the Y chromosome during meiosis.[8] Additionally, 10-25% of human X chromosome genes,[9] and 3-7% of mouse X chromosome genes [10] outside of the PARs show weak expression from the inactive X chromosome.
2. Two-Fold Increased Transcription of a Single ♂ X
Another mechanism common for achieving equal X-related genetic expression between males and females involves two-fold increased transcription of a single male X chromosome. Thus, heterogametic male organisms with one X chromosome may match the level of expression achieved in homogametic females with two active X chromosomes. This mechanism is observed in Drosophila.[11]
The concept of dosage compensation actually originated from an understanding of organisms in which males upregulated X-linked genes two-fold, and was much later extended to account for the observation of the once mysterious Barr bodies. As early as 1932, H.J. Muller carried out a set of experiments which allowed him to track the expression of eye color in flies, which is an X-linked gene. Muller introduced a mutant gene that caused loss of pigmentation in fly eyes, and subsequently noted that males with only one copy of the mutant gene had similar pigmentation to females with two copies of the mutant gene. This led Muller to coin the phrase “dosage compensation” to describe the observed phenomenon of gene expression equalization.[12]
Despite these advances, it was not until Ardhendu Mukherjee and W. Beermann performed more advanced autoradiography experiments in 1965 that scientists could confirm that transcription of genes in the single male X chromosome was double that observed in the two female X chromosomes.[13] Mukherjee and Beermann confirmed this by designing a cellular autoradiography experiment that allowed them to visualize incorporation of [3H]uridine into ribonucleic acid of the X chromosomes. Their studies showed equal levels of [3H]uridine incorporation in the single male X chromosome and the two female X chromosomes. Thus, the investigators concluded that the two-fold increase in the rate of RNA synthesis in the X chromosome of the male relative to those of the female could account for Muller's hypothesized dosage compensation.
In the case of two-fold increased transcription of a single male X chromosome, there is no use for a Barr body, and the male organism must use different genetic machinery to increase the transcriptional output of their single X chromosome. It is common in such organisms for the Y chromosome to be necessary for male fertility, but not for it to play an explicit role in sex determination.[14][15] In Drosophila, for example, the sex lethal (SXL) gene acts as a key regulator of sexual differentiation and maturation in somatic tissue; in XX animals, SXL is activated to repress increased transcription, while in XY animals SXL is inactive and allows male development to proceed via increased transcription of the single X.[15] Several binding sites exist on the Drosophila X chromosome for the dosage compensation complex (DCC), a ribonucleoprotein complex; these binding sites have varying levels of affinity, presumably for varying expression of specific genes.[16] The Male Specific Lethal complex, composed of protein and RNA binds and selectively modifies hundreds of X-linked genes,[17][18] increasing their transcription to levels comparable to female D. melanogaster.
In organisms that use this method of dosage compensation, the presence of one or more X chromosomes must be detected early on in development, as failure to initiate the appropriate dosage compensation mechanisms is lethal.[14] Male specific lethal proteins (MSLs) are a family of four proteins that bind to the X chromosome exclusively in males. The name “MSL” is used because mutations in these genes cause inability to effectively upregulate X-linked genes appropriately, and are thus lethal to males only and not their female counterparts.[14] SXL regulates pre-messenger RNA in males to differentially splice MSLs and result in the appropriate increase in X chromosome transcription observed in male Drosophila. The immediate target of SXL is male specific lethal-2 (MSL-2).[19] Current dogma suggests that the binding of MSL-2 at multiple sites along the SXL gene in females prevents proper MSL-2 translation, and thus, as previously stated, represses the possibility for X-linked genetic upregulation in females. However, all other transcription factors in the MSL family—maleless, MSL-1, and MSL-3—are able to act when SXL is not expressed, as in the case in males. These factors act to increase male X chromosome transcriptional activity. Histone acetylation and the consequent upregulation of X-linked genes in males is dictated by the MSL complex.[20] Specifically, special roX non-coding RNAs on the MSL complexes facilitate binding to the single male X chromosome, and dictate acetylation of specific loci along the X chromosome as well as the formation of euchromatin.[21] Though these RNAs bind at specific sites along the male X chromosome, their effects spread along the length of the chromosome and have the ability to influence large-scale chromatin modifications. The implications of this spreading epigenetic regulation along the male X chromosome is thought to have implications for understanding the transfer of epigenetic activity along long genomic stretches.[11]
3. Decreased Transcription of Both Hermaphroditic Xs by Half
Other species that do not follow the previously discussed conventions of XX females and XY males must find alternative ways to equalize X-linked gene expression among differing sexes. For example, in Caenorhabditis elegans (or C. elegans), sex is determined by the ratio of X chromosomes relative to autosomes;[22] worms with two X chromosomes (XX worms) develop as hermaphrodites, whereas those with only one X chromosome (XO worms) develop as males.[23] This system of sex determination is unique, because there is no male specific chromosome, as is the case in XX/XY sex determination systems. However, as is the case with the previously discussed mechanisms of dosage compensation, failure to express X-linked genes appropriately can still be lethal.[24]
In this XX/XO sex determination system, gene expression on the X chromosome is equalized by downregulating expression of genes on both X chromosomes of hermaphroditic XX organisms by half.[23] In these XX organisms, the dosage compensation complex (DCC) is assembled on both X chromosomes to allow for this tightly regulated change in transcription levels. The DCC is often compared to the condensin complex,[25] which is conserved across the mitotic and meiotic processes of many species. This complex is crucial to the condensation and segregation of chromosomes during both meiosis and mitosis. Because data substantiates the theory that dosage compensation in other species is caused by chromatin-wide modifications, many theorize that the DCC in particular functions similar to the condensin complex in its ability to condense or remodel the chromatin of the X chromosome.[26]
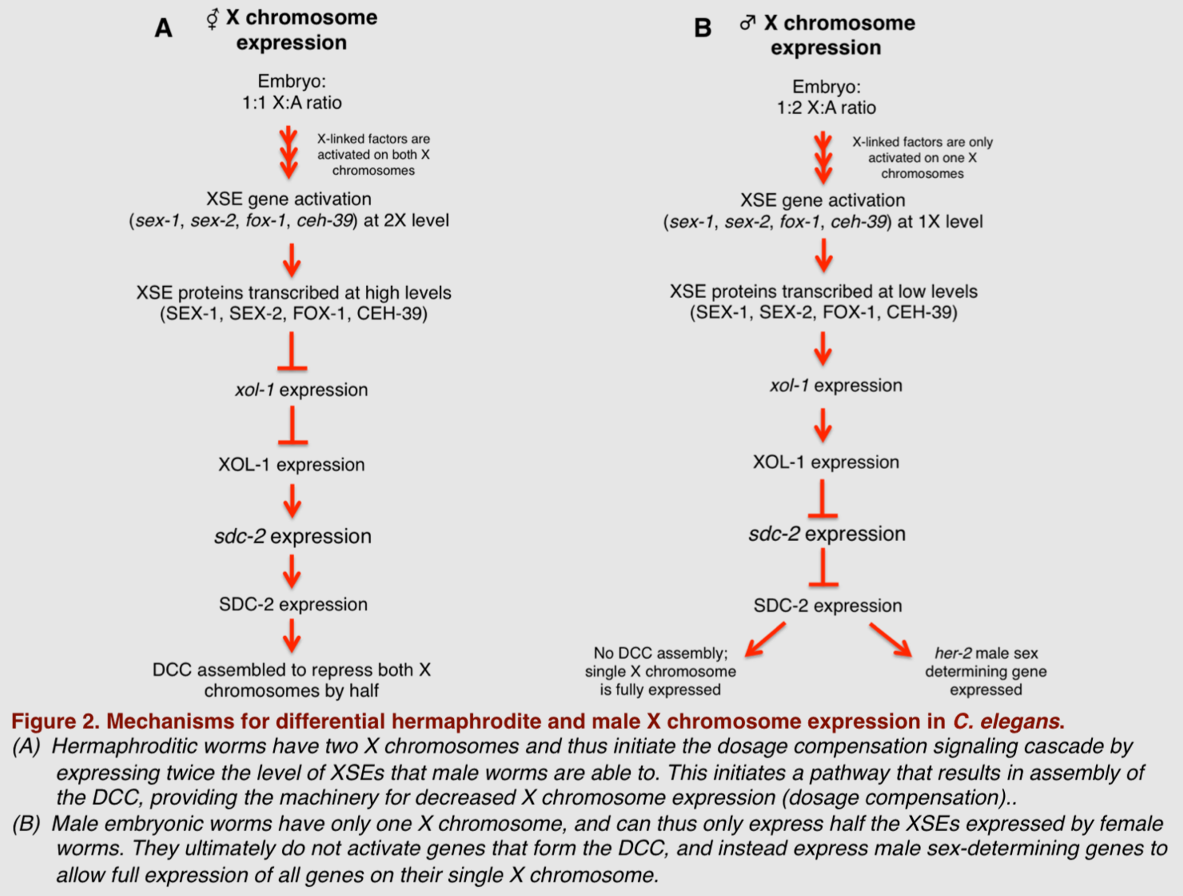
The role of the DCC in this form of dosage compensation was postulated by Barbara J. Meyer in the 1980s, and its individual components and their cooperative function were later parsed out by her lab. Notably, in 1999, data from Meyer's lab showed that SDC-2 is a particularly important transcriptional factor for targeting the DCC to the X chromosome and for assembling DCC components onto the X chromosomes in XX embryos.[27] More recently, Meyer's lab has shown that proteins known as X-linked signal elements (XSEs) operate in concert with SDC-2 to differentially repress and activate other genes in the dosage compensation pathway.[28] By selectively mutating a panel of genes hypothesized to contribute to dosage compensation in worms, Meyer's group demonstrated which XSEs specifically play a role in determining normal dosage compensation. They found that during embryonic development, several X-linked genes—including sex-1, sex-2, fox-1, and ceh-39—act in a combinatorial fashion to selectively repress transcriptional activity of the xol-1 gene in hermaphrodites.[29][30] Xol-1 expression is tightly regulated during early development, and is considered the most upstream gene in sex determination of C. elegans. In fact, xol-1 is often referred to in the literature as the master sex regulatory gene of C. elegans. XX C. elegans embryos have much lower xol-1 expression than their XO counterparts, resulting from overall increases in the amount of SEX-1, SEX-2, CEH-39, and FOX-1 transcription produced in the female embryos. This consequent decrease in xol-1 expression then allows higher SDC-2 expression levels, which aids in the formation and function of the DCC complex in the XX hermaphroditic worms, and in turn results in equalized expression of X-linked genes in the hermaphrodite.
Though all of the above-mentioned XSEs act to reduce xol-1 expression, experimentally reducing expression levels of these individual XSEs has been shown to have a minimal effect on sex determination and successful dosage compensation.[28] This could be in part because these genes encode different proteins that act cooperatively rather than in an isolated fashion; for example, SEX-1 is a nuclear hormone receptor, while FOX-1 is an RNA-binding protein with properties capable of inducing post-transcriptional modifications in the xol-1 target.[28][30][31] However, reducing the level of more than one XSE in different combinational permutations seems to have an additive effect on ensuring proper sex determination and resultant dosage compensation mechanics.[28] This supports the hypothesis that these XSEs act together to achieve the desired sex determination and dosage compensation fate. Thus, in this model organism, the achieved level of X-chromosome expression is directly correlated to the activation of multiple XSEs that ultimately function to repress xol-1 expression in a developing worm embryo. A summary of this C. elegans mechanism of dosage compensation is illustrated below.
4. Other Species-Specific Methods
The ZZ/ZW sex system is used by most birds, as well as some reptiles and insects. In this system the Z is the larger chromosome so the males (ZZ) must silence some genetic material to compensate for the female's (ZW) smaller W chromosome. Instead of silencing the entire chromosome as humans do, male chickens (the model ZZ organism) seem to engage in selective Z silencing, in which they silence only certain genes on the extra Z chromosome.[32][33] Thus, male chickens express an average of 1.4-1.6 of the Z chromosome DNA expressed by female chickens.[34] The Z chromosome expression of male zebra finches and chickens is higher than the autosomal expression rates, whereas X chromosome expression in female humans is equal to autosomal expression rates,[35] illustrating clearly that both male chickens and male zebra finches practice incomplete silencing. Few other ZZ/ZW Systems have been analyzed as thoroughly as the chicken; however a recent study on silkworms [36] revealed similar levels of unequal compensation across male Z chromosomes. Z-specific genes were over-expressed in males when compared to females, and a few genes had equal expression in both male and female Z chromosomes.
In chickens, most of the dosage compensated genes exist on the Zp, or short, arm of the chromosome while the non-compensated genes are on the Zq, or long, arm of the chromosome. The compensated (silenced) genes on Zp resemble a region on the primitive platypus sex chromosome, suggesting an ancestor to the XX/XY system.[37]
4.1. Birds
The sex chromosomes of birds evolved separately from those of mammals and share very little sequence homology with the XY chromosomes.[38] As such, scientists refer to bird sex chromosomes as a ZW sex-determining system, with males possessing two Z chromosomes, and females possessing one Z chromosome and one W. Thus, dosage compensation in birds could be hypothesized to follow a pattern similar to the random X-inactivation observed in most mammals. Alternatively, birds might show decreased transcription of the two Z chromosomes present in the male heterogametic sex, similar to the system observed in the two hermaphrodite X chromosomes of C. elegans. However, bird mechanisms of dosage compensation differ significantly from these precedents. Instead, male birds appear to selectively silence only a few genes along one of their Z chromosomes, rather than randomly silencing one entire Z chromosome.[39] This type of selective silencing has led some people to label birds as “less effective” at dosage compensation than mammals.[35] However, more recent studies have shown that those genes on the Z chromosome which are not inactivated in birds may play an important role in recruiting dosage compensation machinery to the Z chromosome in ZZ organisms.[40] In particular, one of these genes, ScII has been demonstrated to be an ortholog of xol-1, the master sex regulator gene in C. elegans.[40][41] Thus, the function of the selective silencing may be to spare dosage compensation of genes crucial for sex determination of homologous pairing.
While the epigenetic mechanisms behind dosage compensation in birds are poorly understood, especially in comparison to the well-studied mechanisms of dosage compensation in humans and Drosophila, several recent studies have revealed promising sequences. One example is MHM (male hypermethylated) RNA, an Xist-like long noncoding RNA that is expressed only in female chickens (ZW). It is associated with female-specific hyper-acetylation of lysine 16 on histone 4 near the MHM locus on the Z chromosome. This MHM locus is heavily studied as a site of dosage compensation because male Z chromosomes are hypermethylated and thus underexpress genes in this area in comparison to female Z chromosomes which are hyperacetylated and overexpress these genes.[42] There has been debate on whether the MHM locus constitutes dosage compensation, however, since scientists claim that even if the MHM locus has been found to have significantly greater expression in females than in males, it could not even be considered to be a dosage compensation mechanism since it does not balance gene dose between the Z chromosome and autosomes in the heterogametic sex.[43]
Similar to mammals, chickens seem to use CpG islands (segments of Cytosine-phosphate-Guanine that are more readily methylated and silenced than other DNA segments) to regulate gene expression. One study found that CpG islands were found primarily in compensated areas of the Z chromosome—areas that are differentially expressed in male and female chickens. Thus it is likely that these CpG islands are locations where genes on the male Z chromosome are methylated and silenced, but which stay functional on the female Z chromosome.
4.2. Monotremes

Monotremes are a class of basal mammals that also lay eggs.[44] They are an order of mammals that includes platypuses and four species of echidna, all of which are egg-laying mammals. While monotremes use an XX/XY system, unlike other mammals, monotremes have more than two sex chromosomes. The male short-beaked echidna, for example, has nine sex chromosomes—5 Xs and 4 Ys, and the male platypus has 5 Xs and 5 Ys.
Platypuses are a monotreme species whose mechanism of sex determination has been extensively studied. There is some contention in academia about the evolutionary origin and the proper taxonomy of platypuses. A recent study[45] revealed that four platypus X chromosomes, as well as a Y chromosome, are homologous to some regions on the avian Z chromosome. Specifically, platypus X1 shares homology with the chicken Z chromosome, and both share homology with the human chromosome 9. This homology is important when considering the mechanism of dosage compensation in monotremes. In 50% of female platypus cells, only one of the alleles on these X chromosomes is expressed while in the remaining 50% multiple alleles are expressed. This, combined with the portions that are homologous to chicken Z and human 9 chromosomes imply that this level of incomplete silencing may be the ancestral form of dosage compensation.
Regardless of their ambiguous evolutionary history, platypuses have been empirically determined to follow an XY sex-determination system, with females possessing five pairs of X chromosomes as the homogametic sex, and males possessing five X and five Y chromosomes as the heterogametic sex.[46] Because the entire genome of the platypus has yet to be completely sequenced (including one of the X chromosomes),[45] there is still continued investigation as to the definitive mechanism of dosage compensation that Platypuses follow. Research from the laboratory of Jennifer Graves used qPCR and SNP analysis of BACs containing various genes from X chromosomes in order to find whether multiple alleles for particular X-linked genes were being expressed at once, or were otherwise being dosage compensated.[45] Her group found that in female platypuses, some X-linked genes only expressed an allele from one X chromosomes, while other genes expressed multiple alleles.[45] This appears to be a system similar to the selective silencing method of dosage compensation observed in birds. However, about half of all X-linked genes also seemed to stochastically express only one active copy of said gene,[45] alluding to the system of random X-inactivation observed in humans. These findings suggest that platypuses may employ a hybrid form of dosage compensation that combines feature from mammals as well as birds. Understanding the evolution of such a system may have implications for solidifying the true ancestral lineage of monotremes.
4.3. Plants
In addition to humans and flies, some plants also make use of the XX/XY dosage compensation systems. Silene latifolia plants are also either male (XY) or female (XX), with the Y chromosome being smaller, with fewer genes expressed, than the X chromosome. Two separate studies [47] have shown male S. latifolia expression of X-linked genes to be about 70% of the expression in females. If the S. latifolia did not practice dosage compensation, the expected level of X-linked gene expression in males would be 50% that of females, thus the plant practices some degree of dosage compensation but, because male expression is not 100% that of females, it has been suggested that S. latiforia and its dosage compensation system is still evolving. Additionally, in plant species that lack dimorphic sex chromosomes, dosage compensation can occur when aberrant meiotic events or mutations result in either aneuploidy or polyploidy. Genes on the affected chromosome may be upregulated or down-regulated to compensate for the change in the normal number of chromosomes present.
5. X Chromosome Inactivation and Embryonic Stem Cells
XCI is initiated very early during female embryonic development or upon differentiation of female embryonic stem (ES) cells and results in inactivation of one X chromosome in every female somatic cell. This process is initiated very early during development, around the two- to eight-cell stage and is maintained in the developing extra-embryonic tissues of the embryo, including the fetal placenta.[48] Xist RNA induces heterochromatinization of the X chromosome by attracting chromatin modifiers, involved in gene silencing. Xist RNA is tightly associated with the Xi and it is required for X Chromosome Inactivation to occur in cis. Knockout studies in female ES cells and mice have shown that X chromosomes bearing a deletion of the Xist gene are unable to inactivate the mutated X. Most of the human female ES cell lines display an inactivated X chromosome already in the undifferentiated state characterized by XIST expression, XIST coating and accumulated markers of heterochromatin on the Xi.[48]
It is widely thought that human embryos do not employ XCI prior to implantation.[49] Female embryos have an accumulation of Xist RNA on one of the two X chromosomes, beginning around the 8-cell stage. Xist RNA accumulates at the morula and blastocyst stages and is shown to be associated with transcriptional silencing of the Xist-coated chromosomal region, therefore indicating dosage compensation has occurred.[49] Recently, however, it has become increasingly apparent that XCI of the paternal X chromosome is already present from the 4-cell stage onward in all cells of preimplantation mouse embryos, not the 8-cell stages.
6. Xist, Xite, and Tsix and Their Roles in X-Inactivation
Xite and Xist, are both proteins that regulate and facilitate the process of X-inactivation and are important in the silencing of genes within the X chromosome that is being inactivated.[50] These work in combination with Tsix, which is non-coding RNA that is an antisense which downregulates the effects of Xist on the X chromosome in which it is expressed on the maternal X chromosome upon the start regulation of X-inactivation.[51] These three proteins/RNA codons regulate the X-X pair in a cisorientation in order to be able to have both chromosomes available for inhibitory actions. Tsix and Xite have basic protein functions in addition to X-inactivation and regulate the X-X pair in the transorientation. This ensures exclusive silencing for both X chromosomes. Xite and Tsix are both essential within the orientational directional processes in cis and trans as it is seen that without Tsix and Xite in trans it perturbs pairing and counting of genes.[50][51]
Once the Xist is turned off and no longer regulates the process, the Tsix will slowly decrease in expression as well until both proteins’ RNA are no longer being changed by Xic.[51] Xite is the protein that harbors intergenic transcription start sites from hypersensitive sites of allelic crossovers/differences.[50] When X-inactivation begins, the transcription of Xite increases and signals for the downregulation of Tsix in cisorientation, which is on the silent X chromosome, all while promoting the Tsix persistence on the active X chromosome.[52] Xite also has major roles to play in the asymmetry of Tsix expression and generates X chromosome inequality through moving and helping orient the chromosomes to be acted upon by the correct subsequent protein, either Tsix or Xist.[51]
The content is sourced from: https://handwiki.org/wiki/Biology:Sex-Chromosome_Dosage_compensation
References
- Brockdorff, N.; Turner, B.M. (2015). "Dosage compensation in mammals". Cold Spring Harbor Perspectives in Biology 7 (3): a019406. doi:10.1101/cshperspect.a019406. PMID 25731764. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4355265
- Barr, M.L.; Bertram, E.G. (1949). "A morphological distinction between neurones of the male and female, and the behaviour of the nucleolar satellite during accelerated nucleoprotein synthesis". Nature 163 (4148): 676–677. doi:10.1038/163676a0. PMID 18120749. Bibcode: 1949Natur.163..676B. https://dx.doi.org/10.1038%2F163676a0
- Ohno, Susumu (1959). "Sex chromosomes and sex-linked genes". Teratology 4: 111. doi:10.1002/tera.1420040116. https://dx.doi.org/10.1002%2Ftera.1420040116
- Lyon, M.F. (1961). "Gene action in the X-chromosome of the mouse (Mus musculus L.)". Nature 190 (4773): 372–373. doi:10.1038/190372a0. PMID 13764598. Bibcode: 1961Natur.190..372L. https://dx.doi.org/10.1038%2F190372a0
- Ahn, J.; Lee, J. (2008). "X chromosome: X inactivation". Nature Education 1 (1): 24.
- = Beutler, Ernest; Yeh, Mary; Fairbanks, Virgil F. (1962). "The normal human female as a mosaic of X-chromosome activity: studies using the gene for g-6-pd-deficiency as a marker". Proceedings of the National Academy of Sciences of the United States of America 48 (1): 9–16. doi:10.1073/pnas.48.1.9. PMID 13868717. Bibcode: 1962PNAS...48....9B. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=285481
- Beutler, Ernest (2008). "Glucose-6-phosphate dehydrogenase deficiency: a historical perspective". Blood 111: 16–24. doi:10.1182/blood-2007-04-077412. PMID 18156501. https://dx.doi.org/10.1182%2Fblood-2007-04-077412
- Veitia, RA; Veyrunes, F; Bottani, S; Birchler, JA (February 2015). "X chromosome inactivation and active X upregulation in therian mammals: facts, questions, and hypotheses". Journal of Molecular Cell Biology 7 (1): 2–11. doi:10.1093/jmcb/mjv001. PMID 25564545. https://dx.doi.org/10.1093%2Fjmcb%2Fmjv001
- Carrel, L.; Willard, H.F. (2005). "X-inactivation profile reveals extensive variability in X-linked gene expression in females". Nature 434 (7031): 400–4. doi:10.1038/nature03479. PMID 15772666. Bibcode: 2005Natur.434..400C. https://dx.doi.org/10.1038%2Fnature03479
- Berletch, JB; Ma, W; Yang, F; Shendure, J; Noble, WS; Disteche, CM; Deng, X (March 2015). "Escape from X inactivation varies in mouse tissues". PLOS Genetics 11 (3): e1005079. doi:10.1371/journal.pgen.1005079. PMID 25785854. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4364777
- Lucchesi, J.C.; Kuroda, M.I. (2015). "Dosage compensation in Drosophila". Cold Spring Harbor Perspectives in Biology 7 (5): a019398. doi:10.1101/cshperspect.a019398. PMID 25934013. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4448616
- Muller, HJ (1932). "Further studies on the nature and causes of gene mutations". Proc 6th Int Congr Genet 1: 213–255.
- Mukherjee, A.S.; Beermann, W. (1965). "Synthesis of ribonucleic acid by the X-chromosomes of Drosophila melanogaster and the problem of dosage compensation". Nature 207 (4998): 785–786. doi:10.1038/207785a0. PMID 5885936. Bibcode: 1965Natur.207..785M. https://dx.doi.org/10.1038%2F207785a0
- Lucchesi, John C.; Manning, Jerry E. (1987). "Gene dosage compensation in Drosophila melanogaster". Advances in Genetics 24: 371–429. doi:10.1016/S0065-2660(08)60013-9. ISBN 9780120176243. PMID 3124533. https://dx.doi.org/10.1016%2FS0065-2660%2808%2960013-9
- Sass G.L., Pannuti A., Lucchesi J.C. (2003). "Male-specific lethal complex of Drosophila targets activated regions of the X chromosome for chromatin remodeling". Proceedings of the National Academy of Sciences of the United States of America 100 (14): 8287–8291. doi:10.1073/pnas.1332749100. PMID 12829796. Bibcode: 2003PNAS..100.8287S. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=166221
- Dahlsveen, IK; Gilfillan, GD; Shelest, VI; Lamm, R; Becker, PB (February 2006). "Targeting determinants of dosage compensation in Drosophila". PLOS Genetics 2 (2): e5. doi:10.1371/journal.pgen.0020005. PMID 16462942. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1359073
- Zhou Qi (2013). "The epigenome of evolving Drosophila neo-sex chromosomes: dosage compensation and heterochromatin formation". PLOS Biology 11 (11): 1–13. doi:10.1371/journal.pbio.1001711. PMID 24265597. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3825665
- *Deng Xinxian, Meller Victoria H (2009). "Molecularly severe roX1 mutations contribute to dosage compensation in Drosophila". Genesis 47 (1): 49–54. doi:10.1002/dvg.20463. PMID 19101984. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=5029428
- Larschan, E; Alekseyenko, AA; Gortchakov, AA; Peng, S; Li, B; Yang, P; Workman, JL; Park, PJ et al. (12 October 2007). "MSL complex is attracted to genes marked by H3K36 trimethylation using a sequence-independent mechanism". Molecular Cell 28 (1): 121–33. doi:10.1016/j.molcel.2007.08.011. PMID 17936709. https://dx.doi.org/10.1016%2Fj.molcel.2007.08.011
- Meller, VH; Rattner, BP (1 March 2002). "The roX genes encode redundant male-specific lethal transcripts required for targeting of the MSL complex.". The EMBO Journal 21 (5): 1084–91. doi:10.1093/emboj/21.5.1084. PMID 11867536. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=125901
- Maenner, S; Müller, M; Fröhlich, J; Langer, D; Becker, PB (25 July 2013). "ATP-dependent roX RNA remodeling by the helicase maleless enables specific association of MSL proteins". Molecular Cell 51 (2): 174–84. doi:10.1016/j.molcel.2013.06.011. PMID 23870143. https://dx.doi.org/10.1016%2Fj.molcel.2013.06.011
- Meyer BJ. 1997. Sex determination and X chromosome dosage compensation. In C. elegans II (ed. Riddle DL, et al.), pp. 209–240. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Meyer, BJ (2000). "Sex in the wormcounting and compensating X-chromosome dose". Trends in Genetics 16: 247–253. doi:10.1016/s0168-9525(00)02004-7. PMID 10827451. https://dx.doi.org/10.1016%2Fs0168-9525%2800%2902004-7
- Nigon V (1951). "Polyploidie experimentale chez un nematode libre, Rhaditis elegans Maupas". Bulletin Biologique de la France et de la Belgique 85: 187–255.
- Csankovszki, G; Collette, K; Spahl, K; Carey, J; Snyder, M; Petty, E; Patel, U; Tabuchi, T et al. (13 January 2009). "Three distinct condensin complexes control C. elegans chromosome dynamics". Current Biology 19 (1): 9–19. doi:10.1016/j.cub.2008.12.006. PMID 19119011. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2682549
- Eisenmann, David M. (25 June 2005). "Wnt signaling". WormBook: 1–17. doi:10.1895/wormbook.1.7.1. PMID 18050402. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4781570
- Dawes, HE; Berlin, DS; Lapidus, DM; Nusbaum, C; Davis, TL; Meyer, Barbara J. (11 June 1999). "Dosage compensation proteins targeted to X chromosomes by a determinant of hermaphrodite fate". Science 284 (5421): 1800–4. doi:10.1126/science.284.5421.1800. PMID 10364546. https://dx.doi.org/10.1126%2Fscience.284.5421.1800
- Gladden, JM; Meyer, BJ (November 2007). "A ONECUT homeodomain protein communicates X chromosome dose to specify Caenorhabditis elegans sexual fate by repressing a sex switch gene". Genetics 177 (3): 1621–37. doi:10.1534/genetics.106.061812. PMID 17720939. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2147945
- Carmi, Ilil; Kopczynski, Jennifer B.; Meyer, Barbara J. (12 November 1998). "The nuclear hormone receptor SEX-1 is an X-chromosome signal that determines nematode sex". Nature 396 (6707): 168–73. doi:10.1038/24164. PMID 9823896. Bibcode: 1998Natur.396..168C. https://dx.doi.org/10.1038%2F24164
- "Genetic and molecular analysis of fox-1, a numerator element involved in Caenorhabditis elegans primary sex determination". Genetics 151: 617–631. 1999. PMID 9927456. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1460491
- "X-chromosome-counting mechanisms that determine nematode sex". Nature 388 (6638): 200–204. 1997. doi:10.1038/40669. PMID 9217163. Bibcode: 1997Natur.388..200N. https://dx.doi.org/10.1038%2F40669
- Kuroda, Y; Arai, N; Arita, M; Teranishi, M; Hori, T; Harata, M; Mizuno, S (2001). "Absence of Z-chromosome inactivation for five genes in male chickens.". Chromosome Research 9 (6): 457–68. doi:10.1016/s0960-9822(01)00070-7. PMID 11592480. https://dx.doi.org/10.1016%2Fs0960-9822%2801%2900070-7
- McQueen Heather (2001). "Dosage compensation in birds". Current Biology 11 (4): 253–257. doi:10.1016/s0960-9822(01)00070-7. PMID 11592480. https://dx.doi.org/10.1016%2Fs0960-9822%2801%2900070-7
- Ellegren Hans (2007). "Faced with inequality: chicken do not have a general dosage compensation of sex-linked genes". BMC Biology 5: 40. doi:10.1186/1741-7007-5-40. PMID 17883843. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2099419
- Itoh, Y; Melamed, E; Yang, X; Kampf, K; Wang, S; Yehya, N; Van Nas, A; Replogle, K et al. (2007). "Dosage compensation is less effective in birds than in mammals". Journal of Biology 6 (1): 2. doi:10.1186/jbiol53. PMID 17352797. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2373894
- Zha Xingfu (2009). "Dosage analysis of Z chromosome genes using microarray in silkworm, Bombyx mori". Insect Biochemistry and Molecular Biology 29 (5–6): 315–321. doi:10.1016/j.ibmb.2008.12.003. https://dx.doi.org/10.1016%2Fj.ibmb.2008.12.003
- Melamed, E; Arnold, AP (2007). "Regional differences in dosage compensation on the chicken Z chromosome". Genome Biology 8 (9): R202. doi:10.1186/gb-2007-8-9-r202. PMID 17900367. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2375040
- Fridolfsson, AK; Cheng, H; Copeland, NG; Jenkins, NA; Liu, HC; Raudsepp, T; Woodage, T; Chowdhary, B et al. (7 July 1998). "Evolution of the avian sex chromosomes from an ancestral pair of autosomes". Proceedings of the National Academy of Sciences of the United States of America 95 (14): 8147–52. doi:10.1073/pnas.95.14.8147. PMID 9653155. Bibcode: 1998PNAS...95.8147F. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=20944
- Cock A.G. (1964). "Dosage compensation and sex-chromatin in non-mammals". Genet Res Camb 5 (3): 354–365. doi:10.1017/s0016672300034807. https://dx.doi.org/10.1017%2Fs0016672300034807
- McQueen, HA; McBride, D; Miele, G; Bird, AP; Clinton, M (20 February 2001). "Dosage compensation in birds". Current Biology 11 (4): 253–7. doi:10.1016/S0960-9822(01)00070-7. PMID 11250153. https://dx.doi.org/10.1016%2FS0960-9822%2801%2900070-7
- Lieb J.D., Albrecht M.R., Chuan P., Meyer B.J. (1998). "MIX-1: an essential component of the C. elegans mitotic machinery executes X chromosome dosage compensation". Cell 92 (2): 265–277. doi:10.1016/s0092-8674(00)80920-4. PMID 9458050. https://dx.doi.org/10.1016%2Fs0092-8674%2800%2980920-4
- Melamed Esther, Arnold Arthur (2009). "The role of LINEs and CpG islands in dosage compensation on the chicken Z chromosome". Chromosome Research 17 (6): 727–736. doi:10.1007/s10577-009-9068-4. PMID 19672682. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2759020
- Mank, Judith E.; Hosken, David J.; Wedell, Nina (2011-08-01). "Some inconvenient truths about sex chromosome dosage compensation and the potential role of sexual conflict" (in en). Evolution 65 (8): 2133–2144. doi:10.1111/j.1558-5646.2011.01316.x. ISSN 1558-5646. PMID 21790564. https://dx.doi.org/10.1111%2Fj.1558-5646.2011.01316.x
- Charlesworth D., Charlesworth B., Marais G., Charlesworth B., Marais G., Marais G. (2005). "Steps in the evolution of heteromorphic sex chromosomes". Heredity 95 (2): 118–128. doi:10.1038/sj.hdy.6800697. PMID 15931241. https://dx.doi.org/10.1038%2Fsj.hdy.6800697
- Deakin, JE; Hore, TA; Koina, E; Marshall Graves, JA (25 July 2008). "The status of dosage compensation in the multiple X chromosomes of the platypus". PLOS Genetics 4 (7): e1000140. doi:10.1371/journal.pgen.1000140. PMID 18654631. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2453332
- Grützner, F; Graves, JA (December 2004). "A platypus' eye view of the mammalian genome". Current Opinion in Genetics & Development 14 (6): 642–9. doi:10.1016/j.gde.2004.09.006. PMID 15531159. https://dx.doi.org/10.1016%2Fj.gde.2004.09.006
- Meadows, R (2012). "Sex chromosome equality in plants". PLOS Biology 10 (4): e1001312. doi:10.1371/journal.pbio.1001312. PMID 22529748. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3328425
- Barakat, Tahsin Stefan; Gribnau, Joost (2010), Meshorer, Eran; Plath, Kathrin, eds., "X Chromosome Inactivation and Embryonic Stem Cells", The Cell Biology of Stem Cells (Springer US) 695: pp. 132–154, doi:10.1007/978-1-4419-7037-4_10, ISBN 978-1-4419-7036-7 https://dx.doi.org/10.1007%2F978-1-4419-7037-4_10
- van den Berg, Ilse M.; Laven, Joop S.E.; Stevens, Mary; Jonkers, Iris; Galjaard, Robert-Jan; Gribnau, Joost; Hikke van Doorninck, J. (June 2009). "X Chromosome Inactivation Is Initiated in Human Preimplantation Embryos" (in en). The American Journal of Human Genetics 84 (6): 771–779. doi:10.1016/j.ajhg.2009.05.003. PMID 19481196. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2694969
- Ogawa, Yuya; Lee, Jeannie T. (March 2003). "Xite, X-Inactivation Intergenic Transcription Elements that Regulate the Probability of Choice" (in en). Molecular Cell 11 (3): 731–743. doi:10.1016/S1097-2765(03)00063-7. PMID 12667455. https://dx.doi.org/10.1016%2FS1097-2765%2803%2900063-7
- Lee, Jeannie; Davidow, Lance S; Warshawsky, David (April 1999). "Tsix, a gene antisense to Xist at the X-inactivation centre" (in en). Nature Genetics 21 (4): 400–404. doi:10.1038/7734. ISSN 1061-4036. PMID 10192391. http://www.nature.com/articles/ng0499_400.
- Xu, N. (2006-02-24). "Transient Homologous Chromosome Pairing Marks the Onset of X Inactivation" (in en). Science 311 (5764): 1149–1152. doi:10.1126/science.1122984. ISSN 0036-8075. PMID 16424298. Bibcode: 2006Sci...311.1149X. https://dx.doi.org/10.1126%2Fscience.1122984