Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Synaptic dysregulations often result in damaging effects on the central nervous system, resulting in a wide range of brain and neurodevelopment disorders that are caused by mutations disrupting synaptic proteins. Synaptotagmin-1 (SYT1), an identified synaptotagmin protein, plays an essential role in mediating the release of calcium-triggered neurotransmitters (NT) involved in regular synaptic vesicle exocytosis.
- intellectual disability
- synaptic vesicles
- synapse
- neurodevelopment
1. Synaptotagmin Isoforms and SYT1 Function
Synaptic neurotransmission is the fundamental communication between neurons. At the level of synapse, a transmitter is released from one neuron generating a signal on another neuron or target cell that can excites, inhibits, or modulates its cellular activity [1]. The action potential reaching the nerve terminal initiates a rapid influx of Ca2+ions into the postsynaptic cell, causing fusion of vesicles and ultimately the release of neurotransmitters from those vesicles (Figure 1). This release is mediated via the assembly and zippering of SNARE (soluble N-ethylmaleimide-sensitive fusion protein (NSF) attachment protein receptor) complexes that allow the destined membranes to fuse [2]. SNARE complexes in neurons have three components, synaptobrevin or vesicle-associated membrane protein (VAMP), which is the v-SNARE, and two plasma-membrane proteins syntaxin and SNAP-25 (synaptosome-associated protein, 25 kDA) that are the t-SNARES. Once these two membranes combine, they form four-helix bundle aligned in a parallel fashion called the trans-SNARE complex [3]. This machinery is essential for vesicle docking and priming at active zones. Previous studies have indicated the role of synaptotagmin-1 (SYT1) as a dual Ca2+sensor able to carry out both endocytosis and exocytosis independently of one another [4]. These processes are carried out by SYT1’s C2A and C2B domain in a Ca2+dependent manner.
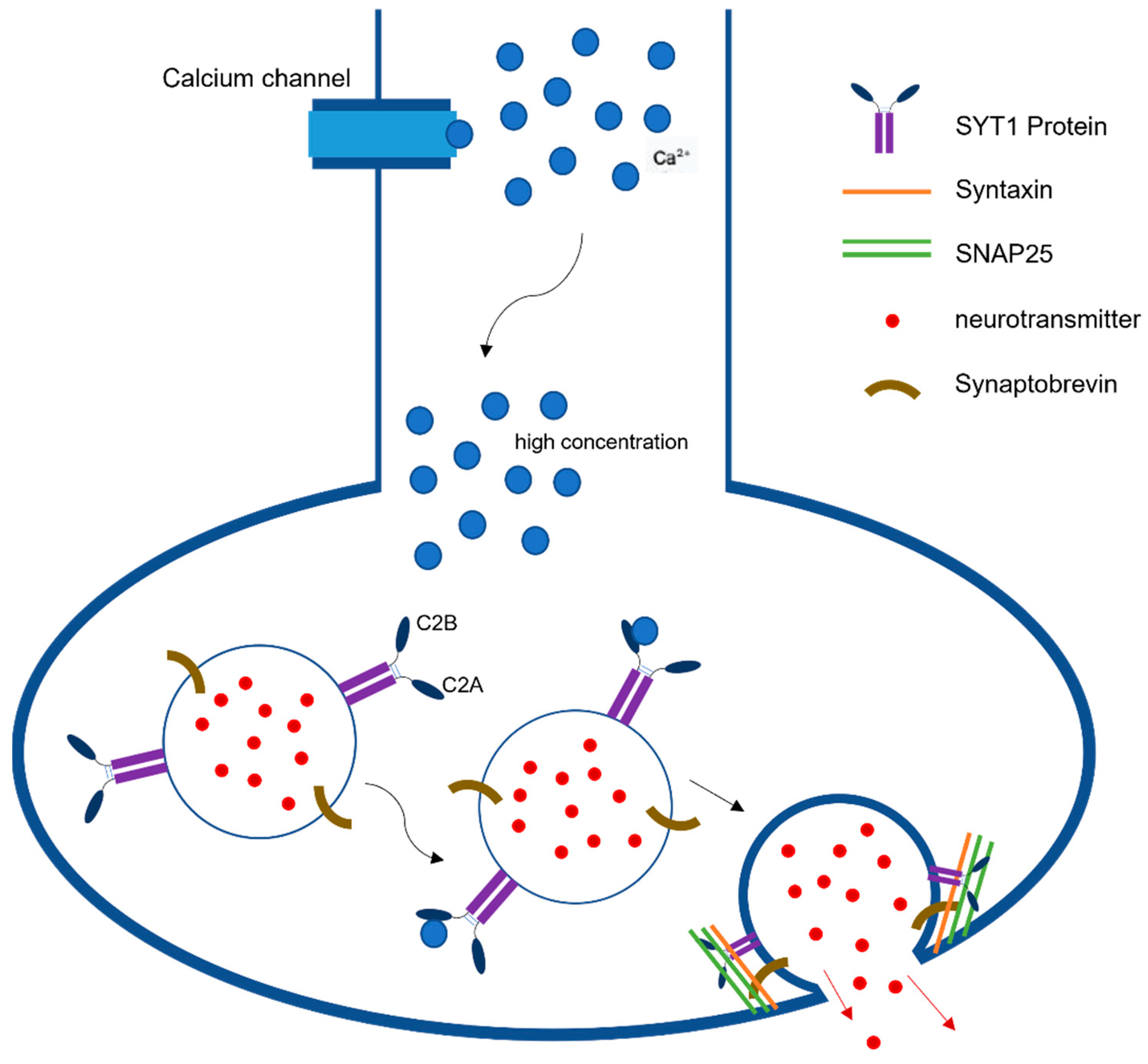
Figure 1. Synaptotagmin-1 Function in Neurotransmission. SYT1 plays a pivotal role in synaptic neurotransmission as a dual Ca2+sensor able to carry out both endocytosis and exocytosis independently. This figure describes the release of neurotransmitters following the detection of high concentrations of Ca2+ by SYT1’s C2B domains.
Synaptotagmins and SNARE proteins play a pivotal role in evoked synchronous neurotransmitter release in a Ca2+ dependent and independent manner. For the purposes of these contents, the SYT isoforms of clinical and functional relevance related to SYT1-associated neurodevelopmental disorder will be discussing (Table 1). These isoforms include SYT2, SYT7, and SYT9 [5][6].
Table 1. Known Synaptotagmin Isoforms, Dysfunctions, and Clinical Mutations. Known synaptotagmin isoforms of clinical and functional relevance related to SYT1-associated neurodevelopmental disorder, including their noted neurotransmitter mechanism of release and related molecular dysfunction.
| Isoform | Location | NT Mechanism of Release | Molecular Dysfunction | Clinical Symptoms of Mutation |
|---|---|---|---|---|
| SYT1 | Synaptic vesicles and secretory granules of neuroendocrine of the hippocampus and forebrain | Independent fast synchronous NT release, clamping mini spontaneous release | Decreased evoked transmission | Developmental delay, sleep disturbances, EEG abnormalities, abnormal motor function, abnormal eye physiology |
| SYT2 | Inhibitory neurons of the brainstem, spinal cord, cerebellum, and striatal neurons, neuromuscular junction | Evoked fast synchronous NT release | Impaired NT vesicle release | Moderate to severe motor deficits, muscle weakness, bulbar deficits, delayed motor development |
| SYT7 | Hippocampus | SV priming and clamping, slow asynchronous NT release | Decrease RRP when KO alongside SYT1, | Mania, behavioral disturbances, bipolar-like behavior (in mice) |
| SYT9 | Dense core vesicles and synaptic vesicles in limbic system and striatum, cortical and hippocampal neurons | Evoked fast synchronous NT release | Decrease in spontaneous miniature rate of synaptic vesicle fusion, inability to initiate membrane fusion | Not Available |
In the forebrain specifically, SYT2 seems to be concentrated in inhibitory neurons, whereas SYT1 dominates excitatory neurons [7]. Interestingly, it has been discovered that KO of SYT2 impairs Ca2+ induced vesicle release and produces severe motor deficits in adolescent mice [7]. In clinical studies, mutations in SYT2 have been reported in the C2B binding domain, similar to SYT1-associated neurodevelopmental disorder, resulting in motor deficits including muscle weakness, bulbar deficits, and delay in motor developmental that ranges from mild to moderate to severe depending on the mutation [8].
SYT9 has the slowest neurotransmitters (NT) release kinetics and lowest affinity for Ca2+ [5][9]. It is concentrated in dense core vesicles and synaptic vesicles within the limbic system and striatum [7][9]. SYT9 is the major isoform responsible for fast-acting synaptic vesicle exocytosis in these neurons [7][9]. In addition to striatal neurons, SYT9 has also been found in cortical and hippocampal neurons It has been demonstrated in previous labs that all three of these neuron classes are regulated by SYT1 and SYT9 but have shown that loss of SYT1 inhibited evoked rapid NT release while the loss of SYT9 had no discernable effect [5][10]. Interestingly, it has been shown that SYT9 can rescue the loss of SYT1 but only when immensely overexpressed [5][7].
Unlike SYT1, C2A is the main Ca2+sensing domain in SYT9 [5][11] and mutations within this domain disrupts the ability for SYT9 to initiate membrane fusion in the presence of AP-driven Ca2+influx [5].
It has been found that SYT7 has redundant function with SYT1 and is capable of SV priming and clamping. It is a high affinity Ca2+sensor for slow asynchronous neurotransmitter release [6][10] and takes part in Ca2+dependent short term synaptic facilitation, SV endocytosis and is responsible for maintaining the RRP of vesicles [10][12]. Simultaneous loss of function of SYT1 and SYT7 demonstrated a decrease in RRP size in both excitatory and inhibitory synapses, whereas loss of function in only one or the other did not show any effect on RRP size [10]. Loss of function of both SYT1 and SYT7 does not seem to alter the rate of vesicle priming into RRP [10]. Due to redundancy in function, phenotypic loss of SYT1 can be partially compensated by SYT7, albeit in immature neurons [6].
SYT1 is responsible for Ca2+dependent and independent fast synchronous NT release [13] and plays a role in clamping mini spontaneous release and in the maintenance of readily releasable pools (RRP) in vesicles. SYT1 is the main isoform found to reside within synaptic vesicles and secretory granules in neurons and neuroendocrine cells located mainly in the hippocampus and forebrain [9]. SYT1 is a low affinity Ca2+sensor that binds to Ca2+following an action potential via the C2B domain initiating the fusion of the SNARE complex thereby providing the main mechanism for spontaneous and synchronous release [6][13][14]. C2B domain is the main site of mutations associated with SYT1-associated neurodevelopmental disorder. Such mutation has demonstrated decreased evoked synchronous release of NTs due to the inhibition of SV fusion [4][13][15]. The precise mechanism will be discussed.
In exocytosis, the SYT1 protein senses the rise in intracellular Ca2+due to an action potential and in turn interacts with negatively charged phospholipids and SNARE proteins to promote membrane fusion [5]. The interaction of SYT1 with negatively charged phospholipid phosphatidylserine is crucial to exocytosis and has been highlighted by previous experiments in C2A mutations that abolished the interaction between SYT1 and phospholipids [16]. However, this mutation in C2A has not had the same effects on the interaction between SYT1 and SNARE proteins [16]. Upon Ca2+binding, SYT1 acts by deforming the plasma membrane or binding to the curved membrane via the C2B domain [13]. This interaction is found to be critical in NT release and exocytosis [13]. Furthermore, when testing the function of SYT1 during exocytosis it was found that C2B mutations greatly disrupted this process whereas corresponding mutations in the C2A domain did not [4].
The efficiency of endocytosis and exocytosis is crucial to establishing efficient neuronal transmission. SYT1 helps to establish this via a bidirectional Ca2+ sensing capability that links endocytosis and exocytosis [17]. Previous labs have recognized that SYT1 is able to initiate slow and small clathrin mediated endocytosis (CME) and/or block bulk-sized endocytosis during increased neural activity [17]. In contrast to the pivotal role of C2B in exocytosis, it has been demonstrated that either C2B or C2A can be involved in SYT1-mediated endocytosis [4]. Interestingly, endocytosis can proceed in both a Ca2+ dependent and independent manner [4]. However, Ca2+binding to either the C2A or C2B domain is necessary in the acceleration of endocytosis [4]. Experiments have shown that mutations in either C2A or C2B (but not in both simultaneously) rescued knockdown-decrease in transferrin uptake, which represented CME and further established the redundancy of C2AB in endocytosis [17]. It was further demonstrated that C2B is needed for bulk endocytosis [17].
2. Mutations in C2AB Inhibit Exocytosis
Previous studies indicate that the most consequential clinical manifestations occur with mutations within the amino acid sequence bind domain of the C2B domain [4][15][18]. The C2B and C2A domain are structurally similar but the C2B domain has a higher affinity for Ca2+-dependent membrane penetration due to the positive cooperativity between C2B and PIP2 [19]. This positive cooperativity promotes the oligomerization of SYT1 [19]. Interestingly, recent studies have shown that both C2B and C2A work in a cooperative manner to assist SYT1 membrane binding. However, C2B still remains the dominant energetic domain in these transmission interactions [16][19][20].
The C2B domain is composed of a series of polylysine residues region at its side that bind to PIP2 in the absence of Ca2+ [21] and two basic residues region located at its bottom; both of which have been shown to be crucial in synchronous NT release [14]. The C2B domain forms a primary and secondary interface with the SNARE complex, which is critical in NT release and exocytosis [13]. A primary interface occurs between the C2B domain and SNARE complex via the interaction between arginine amino acids with glutamates and aspartates between SNAP25 and Syyntaxin-1 [5]. It has been demonstrated that mutations from arginine to glutamines in C2B show decreases in the rate of fusion between C2B and the SNARE complex, thus disrupting exocytosis [13].
Studies have demonstrated that the mutations in C2B domain did not affect the binding rate of Ca2+ to C2B, but rather the time bound decreased [15] and inhibition of NT release via a dominant-negative manner [6][19]. Mutations that neutralized the C2B domain resulted in completely abolishing evoked NT release and disrupt Ca2+ independent vesical docking [14][20]. However, a mutation in C2A resulted in only a 50% decrease in NT release [20]. It is thought that C2B might be the governing domain in evoked NT release due to its increased Ca2+ affinity and stronger membrane binding energy [20]. The mediating role of C2A is not completely understood on a molecular level and must be studied further.
3. Mutations in SYT1 Result in Synaptic Neuronal Transmission Dysfunction
It has been described by Baker et al. [18] that SYT1 mutation causes a dysfunction in synaptic vesicle cycling. In the years since, multiple missense mutations have been implicated in what has been classified as SYT1 -associated neurodevelopmental disorder. Each of the missense mutations causing SYT1associated neurodevelopmental disorder have been implicated in the C2B domain of SYT1 [4][15][18] thereby disrupting SYT1 function. Specifically, it has been demonstrated that mutations reduce the binding between C2B and PIP2 and significantly decrease the membrane sensitivity to Ca2+influx after an AP [21]. On a biomolecular level, this occurs when one of the hydrophobic amino acid residues within the C2B domain are replaced by a neutral or acidic amino acid residue that abolishes Ca2+ and phospholipid binding, which ultimately negatively effects NT release [15][18].
The consequences of these mutations in SYT1 include disrupted vesicle trafficking, mis-localization of SYT1, deficits in evoked synaptic transmission, dose-dependent dominant negative activity, and diminished rate of exocytosis and SV fusion in mammalian synaptic terminals [5][15][18]. However, the severity of the dysfunction depends on the specific mutation. For example, it has been described that while most mutations do impact spontaneous release rates and evoked synchronous NT release, the mutations D303G and I367T in C2B have been witnessed to have a severe impact on evoked synaptic release in comparison to D365E [5]. It has also been observed that the three before-mentioned mutations form unstable complexes with Ca2+, but, interestingly, have increased rapid release of Ca2+, clearly showing that these pathogenic mutations have a reduced Ca2+ sensitivity [5].
In addition, the first mutation identified in a neurodevelopmental disorder, I368T, was shown to decrease rate of SV NT release and rate of evoked SV fusion due to the isoleucine being replaced by threonine. Isoleucine is of particular interest considering that it is a highly conserved residue in the C2B domain that mediates Ca2+ binding [15][18]. Interestingly, previous labs have demonstrated that SYT1 variants had roughly the same level of expression at the nerve terminal as wild type SYT1 except for M303K, which showed decreased expression and more diffuse localization [15].
This entry is adapted from the peer-reviewed paper 10.3390/children9101439
References
- Palay, S.L.; Chan-Palay, V. A Guide to the Synaptic Analysis of the Neuropil. Cold Spring Harb. Sym. Quant. Biol. 1976, 40, 1–16.
- Sollner, T.; Whiteheart, S.W.; Brunner, M.; Erdjument-Bromage, H.; Geromalnos, S.; Tempst, P.; Rothman, J.E. SNAP receptors implicated in vesicle targeting and fusion. Nature 1993, 352, 318–324.
- Han, J.; Pluhackova, K.; Bockmann, R.A. The Multifaceted Role of SNARE Proteins in Membrane Fusion. Front. Physiol. 2017, 8, 5.
- Chapman, E.R. Synaptotagmin: A Ca2+ sensor that triggers exocytosis? Nat. Rev. Mol. Cell Biol. 2002, 3, 498–508.
- Seibert, M.J.; Evans, C.S.; Stanley, K.S.; Wu, Z.; Briguglio, J.S.; Chapman, E.R. Synaptotagmin 9 Modulates Spontaneous Neurotransmitter Release in Striatal Neurons by Regulating Substance P Secretion. bioRxiv 2022. preprint.
- Rosemund, C.; Camacho, M.; Brockmann, M.M.; Zobel, S.; Rosenmund, C. Deconstructing Synaptotagamin-1’s Distinct Roles in Synaptic Vesicle Priming and Neurotransmitter Release. J. Neurosci. 2022, 42, 2856–2871.
- Pang, Z.P.; Melicoff, E.; Padgett, D.; Liu, Y.; Teich, A.F.; Dickey, B.F.; Lin, W.; Adachi, R.; Südhof, T.C. Synaptotagmin-2 Is Essential for Survival and Contributes to Ca2+ Triggering of Neurotransmitter Release in Central and Neuromuscular Synapses. J. Neurosci. 2006, 26, 13493–13504.
- Maselli, R.A.; Wei, D.T.; Hodgson, T.S.; Sampson, J.B.; Bs, J.V.; Smith, H.L.; Pytel, P.; Ferns, M. Dominant and recessive congenital myasthenic syndromes caused by SYT2 mutations. Wiley Muscle Nerve 2021, 10, 219–224.
- Wolfes, A.; Dean, C. The diversity of synaptotagamin isoforms. Sci. Direct 2020, 63, 198–209.
- Bacaj, T.; Wu, D.; Burré, J.; Malenka, R.C.; Liu, X.; Südhof, T.C. Synaptotagmin-1 and -7 Are Redundantly Essential for Maintaining the Capacity of the Readily-Releasable Pool of Synaptic Vesicles. PLoS Biol. 2015, 13, e1002267.
- Südhof, T.C. Synaptotagmins: Why So Many? J. Biol. Chem. 2002, 277, 7629–7632.
- Guan, Z.; Quiñones-Frías, M.C.; Akbergenova, Y.; Littleton, J.T. Drosphila Synaptotagamin 7 negatively regulates synaptic vesicle release and replenishment in a dosage-dependent manner. Elife 2020, 9, e55443.
- Zhou, Q.; Lai, Y.; Bacaj, T.; Zhao, M.; Lyubimov, A.Y.; Uervirojnangkoorn, M.; Zeldin, O.B.; Brewster, A.S.; Sauter, N.K.; Cohen, A.E.; et al. Architecture of the Synaptotagamin-SNARE Machinery for Neuronal Exocytosis. Nature 2015, 525, 62–67.
- Rosamund, C.; Trimbuch, T.; Chang, S. Synaptotagamin-1 drives synchronous Ca2+ triggered fusion bu C2B domain-mediated synaptic vesicle-membrane attachment. Nat. Neurosci. 2018, 21, 33–40.
- Baker, K.; Gordon, S.L.; Melland, H.; Bumbak, F.; Scott, D.; Jiang, T.J.; Owen, D.; Turner, B.J.; Boyd, S.G.; Rossi, M.; et al. SYT1-associated neurodevelopmental disorder: A case series. Brain 2018, 141, 2576–2591.
- Varga, K.; Jiang, Z.J.; Gong, L.W. Phosphatidylserine is critical for vesicle fission during clathrin-mediated endocytosis. J. Neurochem. 2020, 152, 48–60.
- Chen, Y.; Hu, S.; Wu, X.; Xie, Z.; Wang, Y.; Wang, B.; Li, X.; Pei, Y.; Gu, Y.; Wang, C.; et al. Synaptotagamin-1 is bidirectional Ca2+ sensor for neuronal endocytosis. Proc. Natl. Acad. Sci. USA 2022, 119, e2111051119.
- Baker, K.; Gordon, S.L.; Grozeva, D.; Van Kogelenberg, M.; Roberts, N.Y.; Pike, M.; Blair, E.; Hurles, M.E.; Chong, W.K.; Baldeweg, T.; et al. Identification of a human synaptotagmin-1 mutation that perturbs synaptic vesicle cycling. J. Clin. Investig. 2015, 125, 1670–1678.
- Tagliatti, E.; Bello, O.D.; Mendonça, P.R.; Kotzadimitriou, D.; Nicholson, E.; Coleman, J.; Timofeeva, Y.; Rothman, J.E.; Krishnakumar, S.S.; Volynski, K. Synaptotagamin-1 oligomers clamp and regulate different modes of neurotransmitter release. Proc. Natl. Acad. Sci. USA 2020, 117, 3819–3827.
- Gruget, C.; Bello, O.; Coleman, J.; Krishnakumar, S.S.; Perez, E.; Rothman, J.E.; Pincet, F.; Donaldson, S. Synaptotagamin-1 membrane binding is driven by the C2B domain and assisted cooperativity by the C2A domain. Sci. Rep. 2020, 10, 18011.
- Bradberry, M.M.; Courtney, N.A.; Dominguez, M.J.; Lofquist, S.M.; Knox, A.T.; Sutton, R.B.; Chapman, E.R. Molecular Basis for Synaptotagmin-1-Associated Neurodevelopmental Disorder. Neuron 2020, 107, 52–64.
This entry is offline, you can click here to edit this entry!