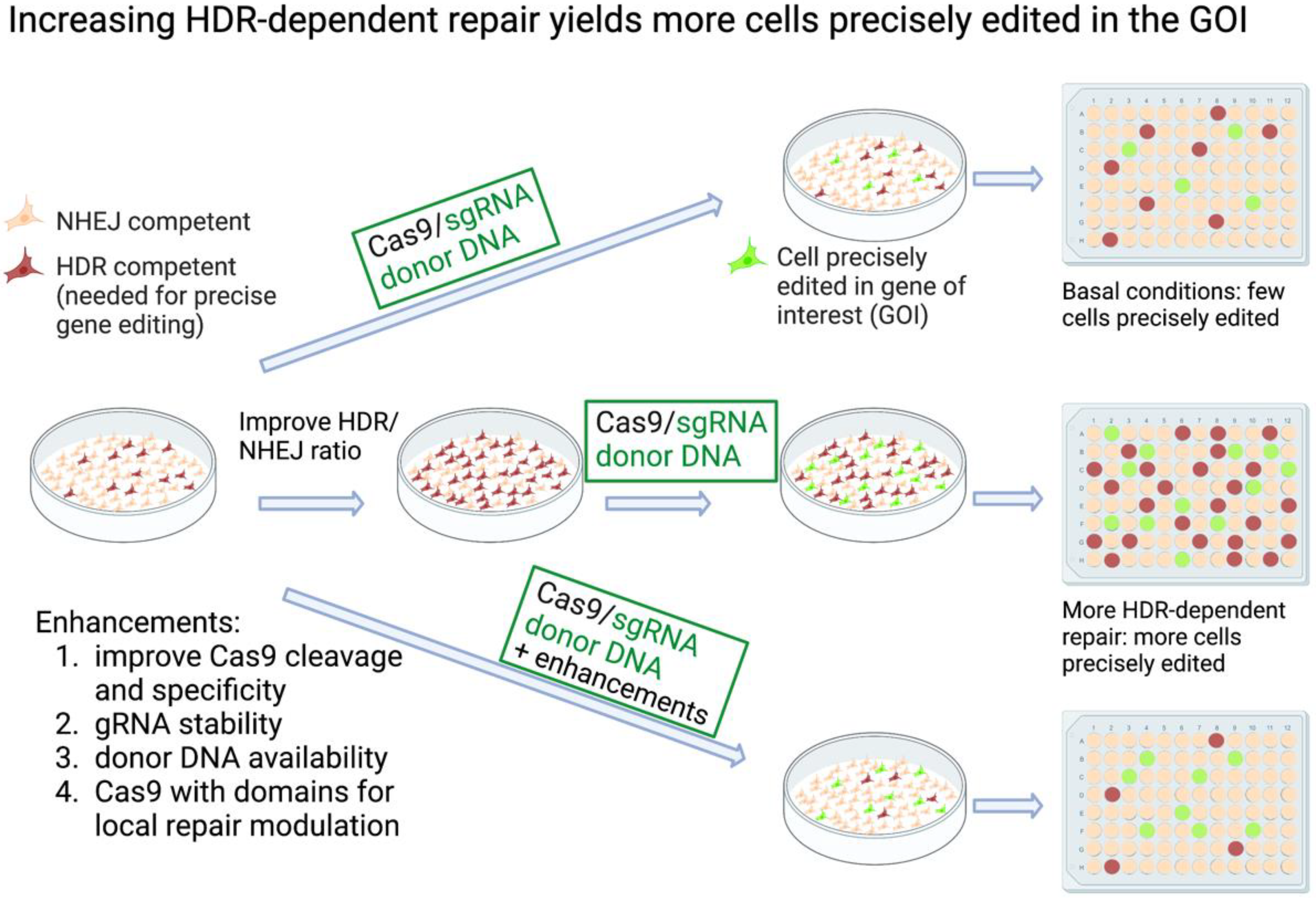
CRISPR (Clustered regularly interspaced short palindromic repeats) technology affords a simple and robust way to edit the genomes of cells, providing powerful tools for basic research and medicine. While using Cas9 to cleave a genomic site is very efficient, making a specific mutation at that site is much less so, as it depends on the endogenous DNA repair machinery. Various strategies have been developed to increase the efficiency of knock-in mutagenesis, mostly focusing on improving homology-directed repair (HDR) while reducing non-homologous end joining (NHEJ). Some approaches affect these repair mechanisms globally, while others target their modulations to the site of the Cas9-induced double-strand break (DSB). Other innovations serve to increase the specificity and the efficiency of the editing mechanisms. In addition, methods such as base editing and prime editing produce knock-in mutations without a DSB.
- CRISPR
- genome editing
- HDR-dependent editing
- scarless selection
1. Introduction
2. Strategies to Increase HDR-Dependent CRISPR-Cas9 Mediated Genome Editing
2.1. Inhibiting NHEJ/Promoting HDR Globally to Increase CRISPR Knock-In Editing
2.2. Improving Cas9 Cleavage and Specificity
2.3. Modifications of the Guide RNA and Donor DNA to Improve Editing Efficiency
2.4. Promoting HDR Specifically at the Break Site
2.5. Editing without a DSB—Circumventing the HDR Requirement
This entry is adapted from the peer-reviewed paper 10.3390/ijms231911919
References
- Barrangou, R.; Doudna, J.A. Applications of CRISPR technologies in research and beyond. Nat. Biotechnol. 2016, 34, 933–941.
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821.
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826.
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823.
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506.
- Takata, M.; Sasaki, M.S.; Sonoda, E.; Morrison, C.; Hashimoto, M.; Utsumi, H.; Yamaguchi-Iwai, Y.; Shinohara, A.; Takeda, S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998, 17, 5497–5508.
- Mjelle, R.; Hegre, S.A.; Aas, P.A.; Slupphaug, G.; Drablos, F.; Saetrom, P.; Krokan, H.E. Cell cycle regulation of human DNA repair and chromatin remodeling genes. DNA Repair 2015, 30, 53–67.
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308.
- Feng, S.; Wang, Z.; Li, A.; Xie, X.; Liu, J.; Li, S.; Li, Y.; Wang, B.; Hu, L.; Yang, L.; et al. Strategies for High-Efficiency Mutation Using the CRISPR/Cas System. Front. Cell Dev. Biol. 2021, 9, 803252.
- Yang, H.; Ren, S.; Yu, S.; Pan, H.; Li, T.; Ge, S.; Zhang, J.; Xia, N. Methods Favoring Homology-Directed Repair Choice in Response to CRISPR/Cas9 Induced-Double Strand Breaks. Int J. Mol. Sci 2020, 21, 6461.
- Yeh, C.D.; Richardson, C.D.; Corn, J.E. Advances in genome editing through control of DNA repair pathways. Nat. Cell Biol. 2019, 21, 1468–1478.
- Sledzinski, P.; Dabrowska, M.; Nowaczyk, M.; Olejniczak, M. Paving the way towards precise and safe CRISPR genome editing. Biotechnol. Adv. 2021, 49, 107737.
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kuhn, R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548.
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542.
- Song, J.; Yang, D.; Xu, J.; Zhu, T.; Chen, Y.E.; Zhang, J. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat. Commun. 2016, 7, 10548.
- Paulsen, B.S.; Mandal, P.K.; Frock, R.L.; Boyraz, B.; Yadav, R.; Upadhyayula, S.; Gutierrez-Martinez, P.; Ebina, W.; Fasth, A.; Kirchhausen, T.; et al. Ectopic expression of RAD52 and dn53BP1 improves homology-directed repair during CRISPR-Cas9 genome editing. Nat. Biomed. Eng. 2017, 1, 878–888.
- Canny, M.D.; Moatti, N.; Wan, L.C.K.; Fradet-Turcotte, A.; Krasner, D.; Mateos-Gomez, P.A.; Zimmermann, M.; Orthwein, A.; Juang, Y.C.; Zhang, W.; et al. Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR-Cas9 genome-editing efficiency. Nat. Biotechnol. 2018, 36, 95–102.
- Shao, S.; Ren, C.; Liu, Z.; Bai, Y.; Chen, Z.; Wei, Z.; Wang, X.; Zhang, Z.; Xu, K. Enhancing CRISPR/Cas9-mediated homology-directed repair in mammalian cells by expressing Saccharomyces cerevisiae Rad52. Int. J. Biochem. Cell Biol. 2017, 92, 43–52.
- Smirnikhina, S.A.; Zaynitdinova, M.I.; Sergeeva, V.A.; Lavrov, A.V. Improving Homology-Directed Repair in Genome Editing Experiments by Influencing the Cell Cycle. Int J. Mol. Sci. 2022, 23, 5992.
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 2014, 3, e04766.
- Rahman, S.H.; Bobis-Wozowicz, S.; Chatterjee, D.; Gellhaus, K.; Pars, K.; Heilbronn, R.; Jacobs, R.; Cathomen, T. The nontoxic cell cycle modulator indirubin augments transduction of adeno-associated viral vectors and zinc-finger nuclease-mediated gene targeting. Hum. Gene Ther. 2013, 24, 67–77.
- Gutschner, T.; Haemmerle, M.; Genovese, G.; Draetta, G.F.; Chin, L. Post-translational Regulation of Cas9 during G1 Enhances Homology-Directed Repair. Cell Rep. 2016, 14, 1555–1566.
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88.
- Lee, J.K.; Jeong, E.; Lee, J.; Jung, M.; Shin, E.; Kim, Y.H.; Lee, K.; Jung, I.; Kim, D.; Kim, S.; et al. Directed evolution of CRISPR-Cas9 to increase its specificity. Nat. Commun. 2018, 9, 3048.
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495.
- Vakulskas, C.A.; Dever, D.P.; Rettig, G.R.; Turk, R.; Jacobi, A.M.; Collingwood, M.A.; Bode, N.M.; McNeill, M.S.; Yan, S.; Camarena, J.; et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018, 24, 1216–1224.
- Maresca, M.; Lin, V.G.; Guo, N.; Yang, Y. Obligate ligation-gated recombination (ObLiGaRe): Custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Res. 2013, 23, 539–546.
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771.
- Tsai, S.Q.; Wyvekens, N.; Khayter, C.; Foden, J.A.; Thapar, V.; Reyon, D.; Goodwin, M.J.; Aryee, M.J.; Joung, J.K. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat. Biotechnol. 2014, 32, 569–576.
- Guilinger, J.P.; Thompson, D.B.; Liu, D.R. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat. Biotechnol. 2014, 32, 577–582.
- Allen, D.; Rosenberg, M.; Hendel, A. Using Synthetically Engineered Guide RNAs to Enhance CRISPR Genome Editing Systems in Mammalian Cells. Front. Genome Ed. 2020, 2, 617910.
- Hendel, A.; Bak, R.O.; Clark, J.T.; Kennedy, A.B.; Ryan, D.E.; Roy, S.; Steinfeld, I.; Lunstad, B.D.; Kaiser, R.J.; Wilkens, A.B.; et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015, 33, 985–989.
- Rahdar, M.; McMahon, M.A.; Prakash, T.P.; Swayze, E.E.; Bennett, C.F.; Cleveland, D.W. Synthetic CRISPR RNA-Cas9-guided genome editing in human cells. Proc. Natl. Acad. Sci. USA 2015, 112, E7110–E7117.
- Lee, K.; Mackley, V.A.; Rao, A.; Chong, A.T.; Dewitt, M.A.; Corn, J.E.; Murthy, N. Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. Elife 2017, 6, e25312.
- Roth, T.L.; Puig-Saus, C.; Yu, R.; Shifrut, E.; Carnevale, J.; Li, P.J.; Hiatt, J.; Saco, J.; Krystofinski, P.; Li, H.; et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 2018, 559, 405–409.
- Shy, B.R.; Vykunta, V.S.; Ha, A.; Talbot, A.; Roth, T.L.; Nguyen, D.N.; Pfeifer, W.G.; Chen, Y.Y.; Blaeschke, F.; Shifrut, E.; et al. High-yield genome engineering in primary cells using a hybrid ssDNA repair template and small-molecule cocktails. Nat. Biotechnol. 2022.
- Tran, N.T.; Bashir, S.; Li, X.; Rossius, J.; Chu, V.T.; Rajewsky, K.; Kuhn, R. Enhancement of Precise Gene Editing by the Association of Cas9 With Homologous Recombination Factors. Front. Genet. 2019, 10, 365.
- Charpentier, M.; Khedher, A.H.Y.; Menoret, S.; Brion, A.; Lamribet, K.; Dardillac, E.; Boix, C.; Perrouault, L.; Tesson, L.; Geny, S.; et al. CtIP fusion to Cas9 enhances transgene integration by homology-dependent repair. Nat. Commun. 2018, 9, 1133.
- Reuven, N.; Adler, J.; Broennimann, K.; Myers, N.; Shaul, Y. Recruitment of DNA Repair MRN Complex by Intrinsically Disordered Protein Domain Fused to Cas9 Improves Efficiency of CRISPR-Mediated Genome Editing. Biomolecules 2019, 9, 584.
- Jayavaradhan, R.; Pillis, D.M.; Goodman, M.; Zhang, F.; Zhang, Y.; Andreassen, P.R.; Malik, P. CRISPR-Cas9 fusion to dominant-negative 53BP1 enhances HDR and inhibits NHEJ specifically at Cas9 target sites. Nat. Commun. 2019, 10, 2866.
- Benitez, E.K.; Lomova Kaufman, A.; Cervantes, L.; Clark, D.N.; Ayoub, P.G.; Senadheera, S.; Osborne, K.; Sanchez, J.M.; Crisostomo, R.V.; Wang, X.; et al. Global and Local Manipulation of DNA Repair Mechanisms to Alter Site-Specific Gene Editing Outcomes in Hematopoietic Stem Cells. Front. Genome Ed. 2020, 2, 601541.
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424.
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157.
- Wolff, J.H.; Haldrup, J.; Thomsen, E.A.; Andersen, S.; Mikkelsen, J.G. piggyPrime: High-Efficacy Prime Editing in Human Cells Using piggyBac-Based DNA Transposition. Front. Genome Ed. 2021, 3, 786893.
- Eggenschwiler, R.; Gschwendtberger, T.; Felski, C.; Jahn, C.; Langer, F.; Sterneckert, J.; Hermann, A.; Luhmann, J.; Steinemann, D.; Haase, A.; et al. A selectable all-in-one CRISPR prime editing piggyBac transposon allows for highly efficient gene editing in human cell lines. Sci. Rep. 2021, 11, 22154.