The tumor microenvironment (TME) is a dynamic network that is created by blood vessels, lymphatic vessels, fibroblasts, immune cells as well as components such as the extracellular matrix (ECM) that establishes a “friendly ecosystem” for cancer cells. Hypoxia induces a cellular adaptive response that elevates the expression of the transcription factors called hypoxia-inducible factors (HIFs) that activate the global gene expression changes in both non-malignant and cancer cells. While most of the studies in this area have focused on the canonical responses to hypoxia, a better understanding is needed for the complex molecular changes that are found in the hypoxic TME. These changes include the deregulation of endoplasmic reticulum (ER) homeostasis, and the subsequent perturbations in protein folding and secretion. The potential for erratic protein folding can also lead to another specialized stress response signaling pathway called the unfolded protein response (UPR). The UPR promotes survival during hypoxia by restoring the endoplasmic and mitochondrial homeostasis, but at times, it can also inhibit the cancer cell’s survival. The maturation of transmembrane and secretory proteins that include proangiogenic receptors and ligands as well as ECM remodeling enzymes takes place in the ER.
- ER-stress
- hypoxia-reoxygenation injury
- TME
1. Hypoxia as an Activator of the UPR in the Tumor Microenvironment
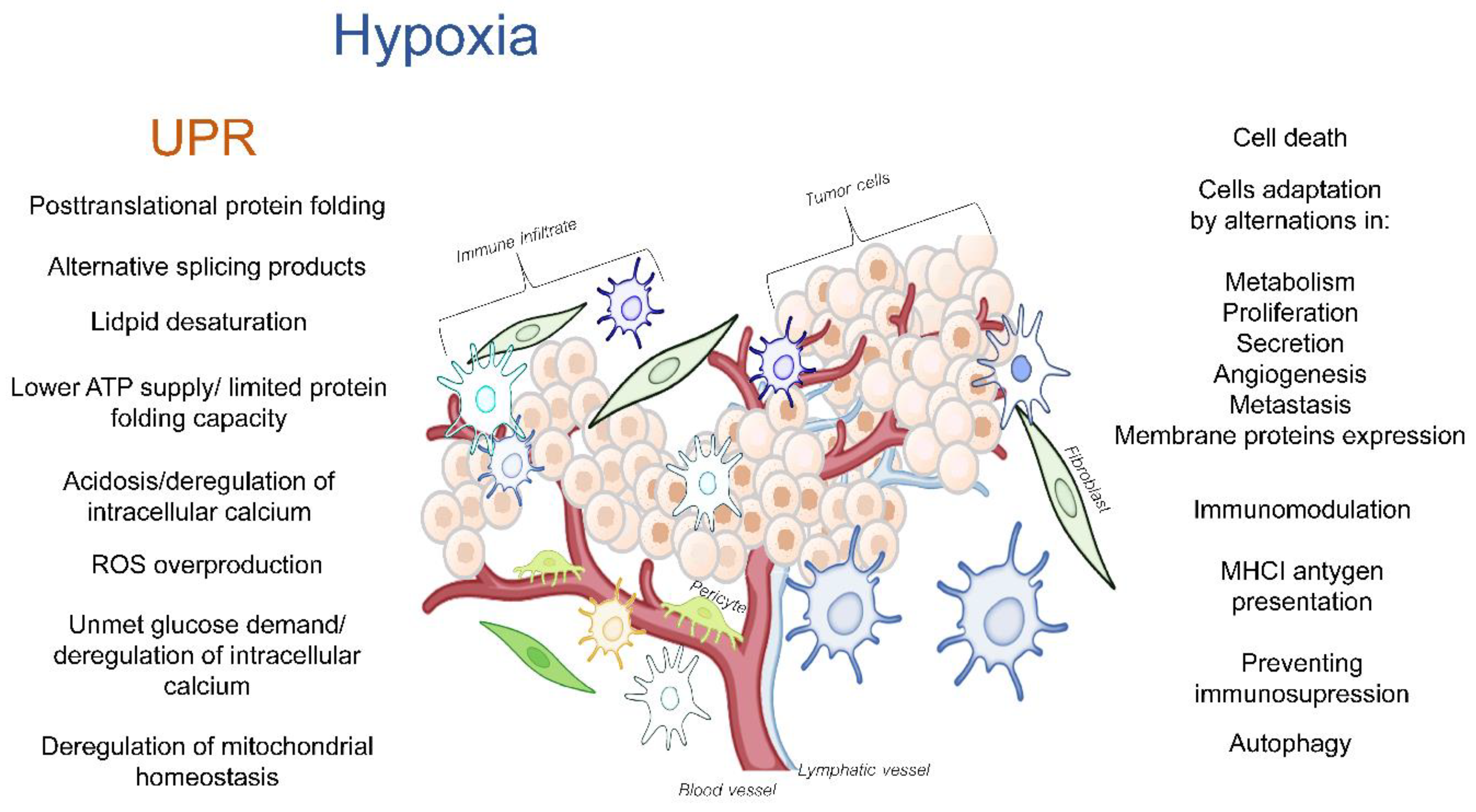
2. The UPR and UPRmt
3. The Crosstalk between Hypoxia and UPR in the TME
4. UPR Activation in TME Cells Depends on Hypoxia Dynamics and Severity
The tumors are extremely heterogenous in terms of their microregions of oxygenation as well as the severity of the hypoxia that ranges from moderate oxygen deprivation to anoxia [14][164][165]. Furthermore, oxygen availably in the TME is often highly dynamic, and it is characterized by the periodic cycling of cells between various levels of oxygenation.
4.1. Anoxia and Extreme Acute Hypoxia
4.2. Moderate and Mild Hypoxia
4.3. Intermittent Hypoxia
This entry is adapted from the peer-reviewed paper 10.3390/cancers14194870
References
- Wouters, B.G.; Koritzinsky, M. Hypoxia signalling through mtor and the unfolded protein response in cancer. Nat. Rev. Cancer 2008, 8, 851–864.
- Bartoszewska, S.; Collawn, J.F. Unfolded protein response (upr) integrated signaling networks determine cell fate during hypoxia. Cell. Mol. Biol. Lett. 2020, 25, 18.
- Manalo, R.V.M. Anastasis and the er stress response: Solving the paradox of the unfolded protein response in cancer. Med. Hypotheses 2017, 109, 25–27.
- Vandewynckel, Y.P.; Laukens, D.; Geerts, A.; Bogaerts, E.; Paridaens, A.; Verhelst, X.; Janssens, S.; Heindryckx, F.; Van Vlierberghe, H. The paradox of the unfolded protein response in cancer. Anticancer Res. 2013, 33, 4683–4694.
- Keith, B.; Simon, M.C. Hypoxia-inducible factors, stem cells, and cancer. Cell 2007, 129, 465–472.
- Heddleston, J.M.; Li, Z.; Lathia, J.D.; Bao, S.; Hjelmeland, A.B.; Rich, J.N. Hypoxia inducible factors in cancer stem cells. Br. J. Cancer 2010, 102, 789–795.
- Koumenis, C. Er stress, hypoxia tolerance and tumor progression. Curr. Mol. Med. 2006, 6, 55–69.
- Kaelin, W.G.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the hif hydroxylase pathway. Mol. Cell 2008, 30, 393–402.
- Jiang, D.D.; Niwa, M.; Koong, A.C. Targeting the ire1 alpha-xbp1 branch of the unfolded protein response in human diseases. Semin. Cancer Biol. 2015, 33, 48–56.
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92.
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219.
- Koumenis, C.; Wouters, B.G. “Translating” tumor hypoxia: Unfolded protein response (upr)-dependent and upr-independent pathways. Mol. Cancer Res. 2006, 4, 423–436.
- Moenner, M.; Pluquet, O.; Bouchecareilh, M.; Chevet, E. Integrated endoplasmic reticulum stress responses in cancer. Cancer Res. 2007, 67, 10631–10634.
- Mujcic, H.; Rzymski, T.; Rouschop, K.M.A.; Koritzinsky, M.; Milani, M.; Harris, A.L.; Wouters, B.G. Hypoxic activation of the unfolded protein response (upr) induces expression of the metastasis-associated gene lamp3. Radiother. Oncol. 2009, 92, 450–459.
- Wang, Y.G.; Alam, G.N.; Ning, Y.; Visioli, F.; Dong, Z.H.; Nor, J.E.; Polverini, P.J. The unfolded protein response induces the angiogenic switch in human tumor cells through the perk/atf4 pathway. Cancer Res. 2012, 72, 5396–5406.
- Rouschop, K.M.A.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes map1lc3b and atg5. J. Clin. Investig. 2010, 120, 127–141.
- Cojocari, D.; Vellanki, R.N.; Sit, B.; Uehling, D.; Koritzinsky, M.; Wouters, B.G. New small molecule inhibitors of upr activation demonstrate that perk, but not ire1 alpha signaling is essential for promoting adaptation and survival to hypoxia. Radiother. Oncol. 2013, 108, 541–547.
- May, D.; Itin, A.; Gal, O.; Kalinski, H.; Feinstein, E.; Keshet, E. Ero1-l alpha plays a key role in a hif-1-mediated pathway to improve disulfide bond formation and vegf secretion under hypoxia: Implication for cancer. Oncogene 2005, 24, 1011–1020.
- Farina, A.R.; Cappabianca, L.; Sebastiano, M.; Zelli, V.; Guadagni, S.; Mackay, A.R. Hypoxia-induced alternative splicing: The 11th hallmark of cancer. J. Exp. Clin. Cancer Res. 2020, 39, 110.
- Young, R.; Ackerman, D.; Quinn, Z.; Mancuso, A.; Gruber, M.; Liu, L.P.; Giannoukos, D.; Bobovnikova-Marjon, E.; Diehl, J.A.; Keith, B.; et al. Dysregulated mTORC1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress. Genes Dev. 2013, 27, 1115–1131.
- Semenza, G.L.; Nejfelt, M.K.; Chi, S.M.; Antonarakis, S.E. Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc. Natl. Acad. Sci. USA 1991, 88, 5680–5684.
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor-1 is a basic-helix-loop-helix-pas heterodimer regulated by cellular o-2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514.
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein vhl targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275.
- Ivan, M.; Kondo, K.; Yang, H.F.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G. Hif alpha targeted for vhl-mediated destruction by proline hydroxylation: Implications for o-2 sensing. Science 2001, 292, 464–468.
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of hif-alpha to the von hippel-lindau ubiquitylation complex by o-2-regulated prolyl hydroxylation. Science 2001, 292, 468–472.
- Mahon, P.C.; Hirota, K.; Semenza, G.L. Fih-1: A novel protein that interacts with hif-1alpha and vhl to mediate repression of hif-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686.
- Bartoszewska, S.; Cabaj, A.; Dabrowski, M.; Collawn, J.F.; Bartoszewski, R. Mir-34c-5p modulates x-box-binding protein 1 (xbp1) expression during the adaptive phase of the unfolded protein response. FASEB J. 2019, 33, 11541–11554.
- Hu, C.-J.; Wang, L.-Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential roles of hypoxia-inducible factor 1alpha (hif-1alpha) and hif-2alpha in hypoxic gene regulation. Mol. Cell. Biol. 2003, 23, 9361–9374.
- Seton-Rogers, S. Hypoxia: Hif switch. Nat. Rev. Cancer 2011, 11, 391.
- Semenza, G.L. Hif-1: Using two hands to flip the angiogenic switch. Cancer Metastasis Rev. 2000, 19, 59–65.
- Koh, M.Y.; Powis, G. Passing the baton: The hif switch. Trends Biochem. Sci. 2012, 37, 364–372.
- Bartoszewski, R.; Moszynska, A.; Serocki, M.; Cabaj, A.; Polten, A.; Ochocka, R.; Dell’Italia, L.; Bartoszewska, S.; Kroliczewski, J.; Dabrowski, M.; et al. Primary endothelial cell-specific regulation of hypoxia-inducible factor (hif)-1 and hif-2 and their target gene expression profiles during hypoxia. FASEB J. 2019, 33, 7929–7941.
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. Hif-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197.
- Huang, D.; Li, T.T.; Li, X.H.; Zhang, L.; Sun, L.C.; He, X.P.; Zhong, X.Y.; Jia, D.Y.; Song, L.B.; Semenza, G.L.; et al. Hif-1-mediated suppression of acyl-coa dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 2014, 8, 1930–1942.
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. Hif-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185.
- Wu, P.F.; Peters, J.M.; Harris, R.A. Adaptive increase in pyruvate dehydrogenase kinase 4 during starvation is mediated by peroxisome proliferator-activated receptor alpha. Biochem. Biophys. Res. Commun. 2001, 287, 391–396.
- Fukuda, R.; Zhang, H.F.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. Hif-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122.
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720.
- Chiche, J.; Brahimi-Horn, M.C.; Pouyssegur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med. 2010, 14, 771–794.
- Chiche, J.; Ilc, K.; Laferriere, J.; Trottier, E.; Dayan, F.; Mazure, N.M.; Brahimi-Horn, M.C.; Pouyssegur, J. Hypoxia-inducible carbonic anhydrase ix and xii promote tumor cell growth by counteracting acidosis through the regulation of the intracellular ph. Cancer Res. 2009, 69, 358–368.
- Chee, N.T.; Lohse, I.; Brothers, S.P. Mrna-to-protein translation in hypoxia. Mol. Cancer 2019, 18, 1–13.
- Thomas, J.D.; Dias, L.M.; Johannes, G.J. Translational repression during chronic hypoxia is dependent on glucose levels. RNA 2008, 14, 771–781.
- Fahling, M. Surviving hypoxia by modulation of mrna translation rate. J. Cell. Mol. Med. 2009, 13, 2770–2779.
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of bnip3 and bnip3l via their bh3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581.
- Yi, T.F.; Papadopoulos, E.; Hagner, P.R.; Wagner, G. Hypoxia-inducible factor-1 alpha (hif-1 alpha) promotes cap-dependent translation of selective mrnas through up-regulating initiation factor eif4e1 in breast cancer cells under hypoxia conditions. J. Biol. Chem. 2013, 288, 18732–18742.
- van den Beucken, T.; Magagnin, M.G.; Jutten, B.; Seigneuric, R.; Lambin, P.; Koritzinsky, M.; Wouters, B.G. Translational control is a major contributor to hypoxia induced gene expression. Radiother. Oncol. 2011, 99, 379–384.
- Papandreou, I.; Lim, A.L.; Laderoute, K.; Denko, N.C. Hypoxia signals autophagy in tumor cells via ampk activity, independent of hif-1, bnip3, and bnip3l. Cell Death Differ. 2008, 15, 1572–1581.
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 2011, 80, 71–99.
- Lodish, H.F. Biogenesis of secretory proteins and cell surface receptors in mammalian cells: Posttranslational modifications and protein folding within the rough endoplasmic reticulum. In Animal Cell Technology; Springer: Dordrecht, The Netherlands, 1997; pp. 15–26.
- Dong, L.; Krewson, E.A.; Yang, L.V. Acidosis activates endoplasmic reticulum stress pathways through gpr4 in human vascular endothelial cells. Int. J. Mol. Sci. 2017, 18, 278.
- Maeyashiki, C.; Melhem, H.; Hering, L.; Baebler, K.; Cosin-Roger, J.; Schefer, F.; Weder, B.; Hausmann, M.; Scharl, M.; Rogler, G.; et al. Activation of ph-sensing receptor ogr1 (gpr68) induces er stress via the ire1 alpha/jnk pathway in an intestinal epithelial cell model. Sci. Rep. 2020, 10, 1438.
- Teixeira, J.; Basit, F.; Swarts, H.G.; Forkink, M.; Oliveira, P.J.; Willems, P.H.G.M.; Koopman, W.J.H. Extracellular acidification induces ros- and mptp-mediated death in hek293 cells. Redox Biol. 2018, 15, 394–404.
- Denzel, M.S.; Antebi, A. Hexosamine pathway and (er) protein quality control. Curr. Opin. Cell Biol. 2015, 33, 14–18.
- Moore, C.E.; Omikorede, O.; Gomez, E.; Willars, G.B.; Herbert, T.P. Perk activation at low glucose concentration is mediated by serca pump inhibition and confers preemptive cytoprotection to pancreatic beta-cells. Mol. Endocrinol. 2011, 25, 315–326.
- Shimizu, Y.; Hendershot, L.M. Oxidative folding: Cellular strategies for dealing with the resultant equimolar production of reactive oxygen species. Antioxid. Redox Signal. 2009, 11, 2317–2331.
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496.
- Verschoor, M.L.; Wilson, L.A.; Singh, G. Mechanisms associated with mitochondrial-generated reactive oxygen species in cancer. Can. J. Physiol. Pharm. 2010, 88, 204–219.
- Gorlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ros: A mutual interplay. Redox Biol. 2015, 6, 260–271.
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2021, 21, 71–88.
- Melber, A.; Haynes, C.M. Uprmt regulation and output: A stress response mediated by mitochondrial-nuclear communication. Cell Res. 2018, 28, 281–295.
- Kueh, H.Y.; Niethammer, P.; Mitchison, T.J. Maintenance of mitochondrial oxygen homeostasis by cosubstrate compensation. Biophys. J. 2013, 104, 1338–1348.
- Shpilka, T.; Haynes, C.M. The mitochondrial upr: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2018, 19, 109–120.
- Munch, C. The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. 2018, 16, 81.
- Jaskiewicz, M.; Moszynska, A.; Serocki, M.; Kroliczewski, J.; Bartoszewska, S.; Collawn, J.F.; Bartoszewski, R. Hypoxia-inducible factor (hif)-3a2 serves as an endothelial cell fate executor during chronic hypoxia. EXCLI J. 2022, 21, 454–469.
- Ratcliffe, P.J. Hif-1 and hif-2: Working alone or together in hypoxia? J. Clin. Investig. 2007, 117, 862–865.
- Loboda, A.; Jozkowicz, A.; Dulak, J. Hif-1 versus hif-2—is one more important than the other? Vasc. Pharmacol. 2012, 56, 245–251.
- Raval, R.R.; Lau, K.W.; Tran, M.G.; Sowter, H.M.; Mandriota, S.J.; Li, J.L.; Pugh, C.W.; Maxwell, P.H.; Harris, A.L.; Ratcliffe, P.J. Contrasting properties of hypoxia-inducible factor 1 (hif-1) and hif-2 in von hippel-lindau-associated renal cell carcinoma. Mol. Cell. Biol. 2005, 25, 5675–5686.
- Zhang, P.; Yao, Q.; Lu, L.; Li, Y.; Chen, P.J.; Duan, C.M. Hypoxia-inducible factor 3 is an oxygen-dependent transcription activator and regulates a distinct transcriptional response to hypoxia. Cell Rep. 2014, 6, 1110–1121.
- Janaszak-Jasiecka, A.; Bartoszewska, S.; Kochan, K.; Piotrowski, A.; Kalinowski, L.; Kamysz, W.; Ochocka, R.J.; Bartoszewski, R.; Collawn, J.F. Mir-429 regulates the transition between hypoxia-inducible factor (hif)1a and hif3a expression in human endothelial cells. Sci. Rep. 2016, 6, 22775.
- Ravenna, L.; Salvatori, L.; Russo, M.A. Hif3alpha: The little we know. FEBS J. 2016, 283, 993–1003.
- Cabaj, A.; Moszynska, A.; Charzynska, A.; Bartoszewski, R.; Dabrowski, M. Functional and hre motifs count analysis of induction of selected hypoxia-responsive genes by hif-1 and hif-2 in human umbilical endothelial cells. Cell. Signal. 2022, 90, 110209.
- Moszynska, A.; Jaskiewicz, M.; Serocki, M.; Cabaj, A.; Crossman, D.K.; Bartoszewska, S.; Gebert, M.; Dabrowski, M.; Collawn, J.F.; Bartoszewski, R. The hypoxia-induced changes in mirna-mrna in rna-induced silencing complexes and hif-2 induced mirnas in human endothelial cells. FASEB J. 2022, 36, e22412.
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luis, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278.
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under er stress and beyond. Nat. Rev. Mol. Cell. Biol. 2012, 13, 89–102.
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789.
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. Er stress induces cleavage of membrane-bound atf6 by the same proteases that process srebps. Mol. Cell 2000, 6, 1355–1364.
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor atf6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799.
- Bartoszewski, R.; Gebert, M.; Janaszak-Jasiecka, A.; Cabaj, A.; Kroliczewski, J.; Bartoszewska, S.; Sobolewska, A.; Crossman, D.K.; Ochocka, R.; Kamysz, W.; et al. Genome-wide mrna profiling identifies rcan1 and gadd45a as regulators of the transitional switch from survival to apoptosis during er stress. FEBS J. 2020, 287, 2923–2947.
- Li, M.Q.; Baumeister, P.; Roy, B.; Phan, T.; Foti, D.; Luo, S.Z.; Lee, A.S. Atf6 as a transcription activator of the endoplasmic reticulum stress element: Thapsigargin stress-induced changes and synergistic interactions with nf-y and yy1. Mol. Cell. Biol. 2000, 20, 5096–5106.
- Zhang, K.Z.; Kaufman, R.J. Signaling the unfolded protein response from the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 25935–25938.
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.P.; Xu, W.H.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. Ire1 alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575.
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting ridd of rna: Ire1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254.
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.Z.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.W.; et al. Xbp1 promotes triple-negative breast cancer by controlling the hif1 alpha pathway. Nature 2014, 508, 103–107.
- Almanza, A.; Mnich, K.; Blomme, A.; Robinson, C.M.; Rodriguez-Blanco, G.; Kierszniowska, S.; McGrath, E.P.; Le Gallo, M.; Pilalis, E.; Swinnen, J.V.; et al. Regulated ire1 alpha-dependent decay (ridd)-mediated reprograming of lipid metabolism in cancer. Nat. Commun. 2022, 13, 2493.
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. Xbp1 mrna is induced by atf6 and spliced by ire1 in response to er stress to produce a highly active transcription factor. Cell 2001, 107, 881–891.
- Bartoszewski, R.; Brewer, J.W.; Rab, A.; Crossman, D.K.; Bartoszewska, S.; Kapoor, N.; Fuller, C.; Collawn, J.F.; Bebok, Z. The unfolded protein response (upr)-activated transcription factor x-box-binding protein 1 (xbp1) induces microrna-346 expression that targets the human antigen peptide transporter 1 (tap1) mrna and governs immune regulatory genes. J. Biol. Chem. 2011, 286, 41862–41870.
- Gebert, M.; Sobolewska, A.; Bartoszewska, S.; Cabaj, A.; Crossman, D.K.; Madanecki, P.; Dabrowski, M.; Collawn, J.F.; Bartoszewski, R.; Kroliczewski, J. Genome-wide mrna profiling identifies x-box-binding protein 1 (xbp1) as an ire1 and puma repressor. Cell. Mol. Life Sci. 2021, 78, 7061–7080.
- Gonen, N.; Sabath, N.; Burge, C.B.; Shalgi, R. Widespread perk-dependent repression of er targets in response to er stress. Sci. Rep. 2019, 9, 4330.
- Han, J.; Backa, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.X.; Yuan, C.L.; Krokowski, D.; Wang, S.Y.; Hatzoglou, M.; et al. Er-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490.
- Rutkowski, D.T.; Kaufman, R.J. All roads lead to atf4. Dev. Cell 2003, 4, 442–444.
- Novoa, I.; Zeng, H.Q.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by gadd34-mediated dephosphorylation of eif2 alpha. J. Cell Biol. 2001, 153, 1011–1021.
- Urano, F.; Wang, X.Z.; Bertolotti, A.; Zhang, Y.H.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the er to activation of jnk protein kinases by transmembrane protein kinase ire1. Science 2000, 287, 664–666.
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652.
- Reimertz, C.; Kogel, D.; Rami, A.; Chittenden, T.; Prehn, J.H.M. Gene expression during er stress-induced apoptosis in neurons: Induction of the bh3-only protein bbc3/puma and activation of the mitochondrial apoptosis pathway. J. Cell Biol. 2003, 162, 587–597.
- Gupta, S.; Giricz, Z.; Natoni, A.; Donnelly, N.; Deegan, S.; Szegezdi, E.; Samali, A. Noxa contributes to the sensitivity of perk-deficient cells to er stress. FEBS Lett. 2012, 586, 4023–4030.
- Rosebeck, S.; Sudini, K.; Chen, T.N.; Leaman, D.W. Involvement of noxa in mediating cellular er stress responses to lytic virus infection. Virology 2011, 417, 293–303.
- Wang, Q.; Mora-Jensen, H.; Weniger, M.A.; Perez-Galan, P.; Wolford, C.; Hai, T.; Ron, D.; Chen, W.P.; Trenkle, W.; Wiestner, A.; et al. Erad inhibitors integrate er stress with an epigenetic mechanism to activate bh3-only protein noxa in cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2200–2205.
- Babour, A.; Bicknell, A.A.; Tourtellotte, J.; Niwa, M. A surveillance pathway monitors the fitness of the endoplasmic reticulum to control its inheritance. Cell 2010, 142, 256–269.
- Sun, M.Y.; Ma, D.S.; Zhao, S.; Wang, L.; Ma, C.Y.; Bai, Y. Salidroside mitigates hypoxia/reoxygenation injury by alleviating endoplasmic reticulum stress-induced apoptosis in h9c2 cardiomyocytes. Mol. Med. Rep. 2018, 18, 3760–3768.
- Tameire, F.; Verginadis, I.I.; Koumenis, C. Cell intrinsic and extrinsic activators of the unfolded protein response in cancer: Mechanisms and targets for therapy. Semin. Cancer Biol. 2015, 33, 3–15.
- Delbrel, E.; Soumare, A.; Naguez, A.; Label, R.; Bernard, O.; Bruhat, A.; Fafournoux, P.; Tremblais, G.; Marchant, D.; Gille, T.; et al. Hif-1 alpha triggers er stress and chop-mediated apoptosis in alveolar epithelial cells, a key event in pulmonary fibrosis. Sci. Rep. 2018, 8, 17939.
- Blazanin, N.; Son, J.; Craig-Lucas, A.B.; John, C.L.; Breech, K.J.; Podolsky, M.A.; Glick, A.B. Er stress and distinct outputs of the ire1 alpha rnase control proliferation and senescence in response to oncogenic ras. Proc. Natl. Acad. Sci. USA 2017, 114, 9900–9905.
- Martin, J. Molecular chaperones and mitochondrial protein folding. J. Bioenerg. Biomembr. 1997, 29, 35–43.
- Baker, B.M.; Nargund, A.M.; Sun, T.; Haynes, C.M. Protective coupling of mitochondrial function and protein synthesis via the eif2 alpha kinase gcn-2. PLoS Genet. 2012, 8, e1002760.
- Binet, F.; Sapieha, P. Er stress and angiogenesis. Cell Metab. 2015, 22, 560–575.
- Sun, L.L.; Chen, C.M.; Zhang, J.; Wang, J.; Yang, C.Z.; Lin, L.Z. Glucose-regulated protein 78 signaling regulates hypoxia-induced epithelial-mesenchymal transition in a549 cells. Front. Oncol. 2019, 9, 137.
- Scheuner, D.; Song, B.B.; McEwen, E.; Liu, C.; Laybutt, R.; Gillespie, P.; Saunders, T.; Bonner-Weir, S.; Kaufman, R.J. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 2001, 7, 1165–1176.
- Koong, A.C.; Auger, E.A.; Chen, E.Y.; Giaccia, A.J. The regulation of grp78 and messenger-rna levels by hypoxia is modulated by protein-kinase-c activators and inhibitors. Radiat. Res. 1994, 138, S60–S63.
- Raiter, A.; Weiss, C.; Bechor, Z.; Ben-Dor, I.; Battler, A.; Kaplan, B.; Hardy, B. Activation of grp78 on endothelial cell membranes by an adam15-derived peptide induces angiogenesis. J. Vasc. Res. 2010, 47, 399–411.
- Koong, A.C.; Chen, E.Y.; Lee, A.S.; Brown, J.M.; Giaccia, A.J. Increased cytotoxicity of chronic hypoxic cells by molecular inhibition of grp78 induction. Int. J. Radiat. Oncol. 1994, 28, 661–666.
- Koumenis, C.; Naczki, C.; Koritzinsky, M.; Rastani, S.; Diehl, A.; Sonenberg, N.; Koromilas, A.; Wouters, B.G. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase perk and phosphorylation of the translation initiation factor eif2alpha. Mol. Cell. Biol. 2002, 22, 7405–7416.
- Blais, J.D.; Filipenko, V.; Bi, M.X.; Harding, H.P.; Ron, D.; Koumenis, C.; Wouters, B.G.; Bell, J.C. Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol. Cell. Biol. 2004, 24, 7469–7482.
- Liu, L.P.; Cash, T.P.; Jones, R.G.; Keith, B.; Thompson, C.B.; Simon, M.C. Hypoxia-induced energy stress regulates mrna translation and cell growth. Mol. Cell 2006, 21, 521–531.
- Ye, J.; Koumenis, C. Atf4, an er stress and hypoxia-inducible transcription factor and its potential role in hypoxia tolerance and tumorigenesis. Curr. Mol. Med. 2009, 9, 411–416.
- Bensellam, M.; Maxwell, E.; Jonas, J.C.; Chan, J.; Laybutt, D.R. Hypoxia induces beta cell death by inhibiting the adaptive upr. Diabetologia 2015, 58, S235.
- Banach, A.; Jiang, Y.P.; Roth, E.; Kuscu, C.; Cao, J.; Lin, R.Z. Cemip upregulates bip to promote breast cancer cell survival in hypoxia. Oncotarget 2019, 10, 4307–4320.
- Fawcett, T.W.; Martindale, J.L.; Guyton, K.Z.; Hai, T.; Holbrook, N.J. Complexes containing activating transcription factor (atf)/camp-responsive-element-binding protein (creb) interact with the ccaat enhancer-binding protein (c/ebp)-atf composite site to regulate gadd153 expression during the stress response. Biochem. J. 1999, 339, 135–141.
- Wolfgang, C.D.; Chen, B.P.C.; Martindale, J.L.; Holbrook, N.J.; Hai, T. Gadd153/chop10, a potential target gene of the transcriptional repressor atf3. Mol. Cell. Biol. 1997, 17, 6700–6707.
- Rouschop, K.M.; Dubois, L.J.; Keulers, T.G.; van den Beucken, T.; Lambin, P.; Bussink, J.; van der Kogel, A.J.; Koritzinsky, M.; Wouters, B.G. Perk/eif2alpha signaling protects therapy resistant hypoxic cells through induction of glutathione synthesis and protection against ros. Proc. Natl. Acad. Sci. USA 2013, 110, 4622–4627.
- Mujcic, H.; Nagelkerke, A.; Rouschop, K.M.A.; Chung, S.; Chaudary, N.; Span, P.N.; Clarke, B.; Milosevic, M.; Sykes, J.; Hill, R.P.; et al. Hypoxic activation of the perk/eif2 alpha arm of the unfolded protein response promotes metastasis through induction of lamp3. Clin. Cancer Res. 2013, 19, 6126–6137.
- Saxena, K.; Jolly, M.K. Acute vs. Chronic vs. Cyclic hypoxia: Their differential dynamics, molecular mechanisms, and effects on tumor progression. Biomolecules 2019, 9, 339.
- Chen, A.; Sceneay, J.; Godde, N.; Kinwel, T.; Ham, S.; Thompson, E.W.; Humbert, P.O.; Moller, A. Intermittent hypoxia induces a metastatic phenotype in breast cancer. Oncogene 2018, 37, 4214–4225.
- Kochan-Jamrozy, K.; Kroliczewski, J.; Moszynska, A.; Collawn, J.F.; Bartoszewski, R. Mirna networks modulate human endothelial cell adaptation to cyclic hypoxia. Cell. Signal. 2019, 54, 150–160.
- Ivanova, I.G.; Park, C.V.; Yemm, A.I.; Kenneth, N.S. Perk/eif2 alpha signaling inhibits hif-induced gene expression during the unfolded protein response via yb1-dependent regulation of hif1 alpha translation. Nucleic Acids Res. 2018, 46, 3878–3890.
- Hiwatashi, Y.; Kanno, K.; Takasaki, C.; Goryo, K.; Sato, T.; Torii, S.; Sogawa, K.; Yasumoto, K. Phd1 interacts with atf4 and negatively regulates its transcriptional activity without prolyl hydroxylation. Exp. Cell Res. 2011, 317, 2789–2799.
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The role of the perk/eif2 alpha/atf4/chop signaling pathway in tumor progression during endoplasmic reticulum stress. Curr. Mol. Med. 2016, 16, 533–544.
- Yang, D.; Gao, L.L.; Wang, T.F.; Qiao, Z.D.; Liang, Y.J.; Zhang, P. Hypoxia triggers endothelial endoplasmic reticulum stress and apoptosis via induction of vldl receptor. FEBS Lett. 2014, 588, 4448–4456.
- Xie, P.; Duan, Y.C.; Guo, X.Z.; Hu, L.N.; Yu, M.H. Sala attenuates hypoxia-induced endothelial endoplasmic reticulum stress and apoptosis via down-regulation of vldl receptor expression. Cell. Physiol. Biochem. 2015, 35, 17–28.
- Loinard, C.; Zouggari, Y.; Rueda, P.; Ramkhelawon, B.; Cochain, C.; Vilar, J.; Recalde, A.; Richart, A.; Charue, D.; Duriez, M.; et al. C/ebp homologous protein-10 (chop-10) limits postnatal neovascularization through control of endothelial nitric oxide synthase gene expression. Circulation 2012, 125, 1014–1026.
- Gebert, M.; Bartoszewska, S.; Janaszak-Jasiecka, A.; Moszynska, A.; Cabaj, A.; Kroliczewski, J.; Madanecki, P.; Ochocka, R.J.; Crossman, D.K.; Collawn, J.F.; et al. Piwi proteins contribute to apoptosis during the upr in human airway epithelial cells. Sci. Rep. 2018, 8, 16431.
- De Pascali, F.; Hemann, C.; Samons, K.; Chen, C.A.; Zweier, J.L. Hypoxia and reoxygenation induce endothelial nitric oxide synthase uncoupling in endothelial cells through tetrahydrobiopterin depletion and s-glutathionylation. Biochemistry 2014, 53, 3679–3688.
- Badran, M.; Abuyassin, B.; Golbidi, S.; Ayas, N.; Laher, I. Uncoupling of vascular nitric oxide synthase caused by intermittent hypoxia. Oxid. Med. Cell Longev. 2016, 2016, 2354870.
- Jeong, K.; Kim, K.; Kim, H.; Oh, Y.; Kim, S.J.; Jo, Y.; Choe, W. Hypoxia induces cyclophilin b through the activation of transcription factor 6 in gastric adeno carcinoma cells. Oncol. Lett. 2015, 9, 2854–2858.
- Sicari, D.; Fantuz, M.; Bellazzo, A.; Valentino, E.; Apollonio, M.; Pontisso, I.; Di Cristino, F.; Dal Ferro, M.; Bicciato, S.; Del Salf, G.; et al. Mutant p53 improves cancer cells’ resistance to endoplasmic reticulum stress by sustaining activation of the upr regulator atf6. Oncogene 2019, 38, 6184–6195.
- Xiao, W.; Cao, R.C.; Yang, W.J.; Tan, J.H.; Liu, R.Q.; Kan, H.P.; Zhou, L.; Zhang, N.; Chen, Z.Y.; Chen, X.M.; et al. Roles and clinical significances of atf6, emc6, and apaf1 in prognosis of pancreatic cancer. Front. Genet. 2022, 12, 730847.
- Liang, H.; Zhou, Z.; Chen, C. Abstract 2042: Hypoxia induces mir-153 through the ire1α-xbp1 pathway to fine-tune the hif1α/vegfa axis in breast cancer angiogenesis. Cancer Res. 2018, 78, 2042.
- Liang, H.; Xiao, J.; Zhou, Z.; Wu, J.; Ge, F.; Li, Z.; Zhang, H.; Sun, J.; Li, F.; Liu, R.; et al. Hypoxia induces mir-153 through the ire1alpha-xbp1 pathway to fine tune the hif1alpha/vegfa axis in breast cancer angiogenesis. Oncogene 2018, 37, 1961–1975.
- Xu, X.D.; Qimuge, A.D.; Wang, H.L.; Xing, C.; Gu, Y.; Liu, S.S.; Xu, H.; Hu, M.R.; Song, L. Ire1 alpha/xbp1s branch of upr links hif1 alpha activation to mediate angii-dependent endothelial dysfunction under particulate matter (pm) 2.5 exposure. Sci. Rep. 2017, 7, 13507.
- Romero-Ramirez, L.; Cao, H.; Nelson, D.; Hammond, E.; Lee, A.H.; Yoshida, H.; Mori, K.; Glimcher, L.H.; Denko, N.C.; Giaccia, A.J.; et al. Xbp1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004, 64, 5943–5947.
- Romero, L.; Cao, H.; Hammond, E.; Giaccia, A.J.; Le, Q.T.; Koong, A.C. Xbp1 is essential for survival under hypoxic conditions and is required for tumor growth. Int. J. Radiat. Oncol. 2004, 60, S192–S193.
- Bouchecareilh, M.; Chevet, E.; Bikfalvi, A.; Moenner, M.; Drogat, B.; Auguste, P.; Nguyen, D.T.; Pineau, R.; Nalbantoglu, J.; Kaufman, R. Ire1 signaling is essential for ischemia-induced vascular endothelial growth factor-a expression and contributes to angiogenesis and tumor growth in vivo. B Cancer 2007, 94, S283–S284.
- Drogat, B.; Auguste, P.; Nguyen, D.T.; Bouchecareilh, M.; Pineau, R.; Nalbantoglu, J.; Kaufman, R.J.; Chevet, E.; Bikfalvi, A.; Moenner, M. Ire1 signaling is essential for ischemia-induced vascular endothelial growth factor-a expression and contributes to angiogenesis and tumor growth in vivo. Cancer Res. 2007, 67, 6700–6707.
- Karar, J.; Dolt, K.S.; Pasha, M.A.Q. Endoplasmic reticulum stress response in murine kidney exposed to acute hypobaric hypoxia. FEBS Lett. 2008, 582, 2521–2526.
- Karali, E.; Bellou, S.; Stellas, D.; Klinakis, A.; Murphy, C.; Fotsis, T. Vegf signals through atf6 and perk to promote endothelial cell survival and angiogenesis in the absence of er stress. Mol. Cell 2014, 54, 559–572.
- Urra, H.; Hetz, C. A novel er stress-independent function of the upr in angiogenesis. Mol. Cell 2014, 54, 542–544.
- Ghosh, R.; Lipson, K.L.; Sargent, K.E.; Mercurio, A.M.; Hunt, J.S.; Ron, D.; Urano, F. Transcriptional regulation of vegf-a by the unfolded protein response pathway. PLoS ONE 2010, 5, e9575.
- Pereira, E.R.; Liao, N.; Neale, G.A.; Hendershot, L.M. Transcriptional and post-transcriptional regulation of proangiogenic factors by the unfolded protein response. PLoS ONE 2010, 5, e12521.
- Roybal, C.N.; Hunsaker, L.A.; Barbash, O.; Vander Jagt, D.L.; Abcouwer, S.F. The oxidative stressor arsenite activates vascular endothelial growth factor mrna transcription by an atf4-dependent mechanism. J. Biol. Chem. 2005, 280, 20331–20339.
- Kyriakakis, E.; Philippova, M.; Joshi, M.B.; Pfaff, D.; Bochkov, V.; Afonyushkin, T.; Erne, P.; Resink, T.J. T-cadherin attenuates the perk branch of the unfolded protein response and protects vascular endothelial cells from endoplasmic reticulum stress-induced apoptosis. Cell. Signal. 2010, 22, 1308–1316.
- Afonyushkin, T.; Oskolkova, O.V.; Philippova, M.; Resink, T.J.; Erne, P.; Binder, B.R.; Bochkov, V.N. Oxidized phospholipids regulate expression of atf4 and vegf in endothelial cells via nrf2-dependent mechanism: Novel point of convergence between electrophilic and unfolded protein stress pathways. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1007–1013.
- Liu, L.; Qi, X.; Chen, Z.; Shaw, L.; Cai, J.; Smith, L.H.; Grant, M.B.; Boulton, M.E. Targeting the ire1alpha/xbp1 and atf6 arms of the unfolded protein response enhances vegf blockade to prevent retinal and choroidal neovascularization. Am. J. Pathol. 2013, 182, 1412–1424.
- Longchamp, A.; Mirabella, T.; Arduini, A.; MacArthur, M.R.; Das, A.; Trevino-Villarreal, J.H.; Hine, C.; Ben-Sahra, I.; Knudsen, N.H.; Brace, L.E.; et al. Amino acid restriction triggers angiogenesis via gcn2/atf4 regulation of vegf and h2s production. Cell 2018, 173, 117–129.e114.
- Terashima, J.; Tachikawa, C.; Kudo, K.; Habano, W.; Ozawa, S. An aryl hydrocarbon receptor induces vegf expression through atf4 under glucose deprivation in hepg2. BMC Mol. Biol. 2013, 14, 27.
- Pollreisz, A.; Afonyushkin, T.; Oskolkova, O.V.; Gruber, F.; Bochkov, V.N.; Schmidt-Erfurth, U. Retinal pigment epithelium cells produce vegf in response to oxidized phospholipids through mechanisms involving atf4 and protein kinase ck2. Exp. Eye Res. 2013, 116, 177–184.
- Chai, L.; Ling, K.; He, X.; Yang, R. Expression of atf4 and vegf in chorionic villus tissue in early spontaneous abortion. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 170, 434–438.
- Oskolkova, O.V.; Afonyushkin, T.; Leitner, A.; von Schlieffen, E.; Gargalovic, P.S.; Lusis, A.J.; Binder, B.R.; Bochkov, V.N. Atf4-dependent transcription is a key mechanism in vegf up-regulation by oxidized phospholipids: Critical role of oxidized sn-2 residues in activation of unfolded protein response. Blood 2008, 112, 330–339.
- Pereira, E.R.; Frudd, K.; Awad, W.; Hendershot, L.M. Endoplasmic reticulum (er) stress and hypoxia response pathways interact to potentiate hypoxia-inducible factor 1 (hif-1) transcriptional activity on targets like vascular endothelial growth factor (vegf). J. Biol. Chem. 2014, 289, 3352–3364.
- Chiang, C.K.; Nangaku, M.; Tanaka, T.; Iwawaki, T.; Inagi, R. Endoplasmic reticulum stress signal impairs erythropoietin production: A role for atf4. Am. J. Physiol.-Cell Physiol. 2013, 304, C342–C353.
- Bartoszewski, R.; Kroliczewski, J.; Piotrowski, A.; Jasiecka, A.J.; Bartoszewska, S.; Vecchio-Pagan, B.; Fu, L.; Sobolewska, A.; Matalon, S.; Cutting, G.R.; et al. Codon bias and the folding dynamics of the cystic fibrosis transmembrane conductance regulator. Cell. Mol. Biol. Lett. 2016, 21, 23.
- Sakaki, K.; Kaufman, R.J. Interaction between quality control systems for er protein folding and rna biogenesis. Worm 2013, 2, e23005.
- Araki, K.; Nagata, K. Protein folding and quality control in the er. Cold Spring Harb. Perspect. Biol. 2012, 4, a015438.
- Hebert, D.N.; Molinari, M. In and out of the er: Protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007, 87, 1377–1408.
- Listowski, M.A.; Heger, E.; Boguslawska, D.M.; Machnicka, B.; Kuliczkowski, K.; Leluk, J.; Sikorski, A.F. Micrornas: Fine tuning of erythropoiesis. Cell. Mol. Biol. Lett. 2013, 18, 34–46.
- Evans, S.M.; Koch, C.J. Prognostic significance of tumor oxygenation in humans. Cancer Lett. 2003, 195, 1–16.
- Le, Q.T.; Denko, N.C.; Giaccia, A.J. Hypoxic gene expression and metastasis. Cancer Metast. Rev. 2004, 23, 293–310.
- Durand, R.E.; Aquino-Parsons, C. Clinical relevance of intermittent tumour blood flow. Acta Oncol. 2001, 40, 929–936.
- Lanzen, J.; Braun, R.D.; Klitzman, B.; Brizel, D.; Secomb, T.W.; Dewhirst, M.W. Direct demonstration of instabilities in oxygen concentrations within the extravascular compartment of an experimental tumor. Cancer Res. 2006, 66, 2219–2223.
- Koritzinsky, M.; Magagnin, M.G.; van den Beucken, T.; Seigneuric, R.; Savelkouls, K.; Dostie, J.; Pyronnet, S.; Kaufman, R.J.; Weppler, S.A.; Voncken, J.W.; et al. Gene expression during acute and prolonged hypoxia is regulated by distinct mechanisms of translational control. EMBO J. 2006, 25, 1114–1125.
- Chaplin, D.J.; Hill, S.A. Temporal heterogeneity in microregional erythrocyte flux in experimental solid tumors. Br. J. Cancer 1995, 71, 1210–1213.
- Sutherland, R.M.; Ausserer, W.A.; Murphy, B.J.; Laderoute, K.R. Tumor hypoxia and heterogeneity: Challenges and opportunities for the future. Semin. Radiat. Oncol. 1996, 6, 59–70.
- Cardenas-Navia, L.I.; Yu, D.H.; Braun, R.D.; Brizel, D.M.; Secomb, T.W.; Dewhirst, M.W. Tumor-dependent kinetics of partial pressure of oxygen fluctuations during air and oxygen breathing. Cancer Res. 2004, 64, 6010–6017.
- Janssen, H.L.K.; Haustermans, K.M.G.; Sprong, D.; Blommestijn, G.; Hofland, I.; Hoebers, F.J.; Blijweert, E.; Raleigh, J.A.; Semenza, G.L.; Varia, M.A.; et al. Hif-1a, pimonidazole, and iododeoxyuridine to estimate hypoxia and perfusion in human head-and-neck tumors. Int. J. Radiat. Oncol. 2002, 54, 1537–1549.
- Bader, S.B.; Dewhirst, M.W.; Hammond, E.M. Cyclic hypoxia: An update on its characteristics, methods to measure it and biological implications in cancer. Cancers 2021, 13, 23.
- Zepeda, A.B.; Pessoa, A.; Castillo, R.L.; Figueroa, C.A.; Pulgar, V.M.; Farias, J.G. Cellular and molecular mechanisms in the hypoxic tissue: Role of hif-1 and ros. Cell Biochem. Funct. 2013, 31, 451–459.
- Hsieh, C.H.; Shyu, W.C.; Chiang, C.Y.; Kuo, J.W.; Shen, W.C.; Liu, R.S. Nadph oxidase subunit 4-mediated reactive oxygen species contribute to cycling hypoxia-promoted tumor progression in glioblastoma multiforme. PLoS ONE 2011, 6, e23945.