CRISPR technology affords a simple and robust way to edit the genomes of cells, providing powerful tools for basic research and medicine. While using Cas9 to cleave a genomic site is very efficient, making a specific mutation at that site is much less so, as it depends on the endogenous DNA repair machinery. Various strategies have been developed to increase the efficiency of knock-in mutagenesis, mostly focusing on improving homology-directed repair (HDR) while reducing non-homologous end joining (NHEJ). Some approaches affect these repair mechanisms globally, while others target their modulations to the site of the Cas9-induced double-strand break (DSB). Other innovations serve to increase the specificity and the efficiency of the editing mechanisms. In addition, methods such as base editing and prime editing produce knock-in mutations without a DSB.
1. Introduction
CRISPR (Clustered regularly interspaced short palindromic repeats) is an elegant system for making DNA double-strand breaks (DSB) in a highly efficient and specific manner. Originating in the bacterial world, where it functions to protect bacteria from invading phages, the system has been leveraged to provide the first step in the genome engineering of organisms from microbes to humans [
1,
2,
3,
4]. The most commonly used components of the system are Cas9, having endonuclease functions, and the associated guide RNA (gRNA), which dictates where the Cas9 will bind and cleave the DNA. To simplify the system, the original two-part Cas9-associated RNAs, the CRISPR RNA (crRNA) and trans-activating CRISPR RNA (tracrRNA), are expressed as a fused single guide RNA (sgRNA), having both the programmable part for identifying the targeted locus and the tracrRNA portion for association with the Cas9 [
2].
CRISPR technology provides a very efficient and easy-to-use method for targeting a specific locus in the genome. However, since editing of that locus depends on the endogenous DNA repair pathways, making a specific edit (a “knock-in” mutation) is much less efficient than using CRISPR to make a functional knock-out. Repair of a DSB by non-homologous end joining (NHEJ) often results in small insertions or deletions (indels) at the site of the break (reviewed in [
5]). This type of repair can lead to a frameshift, resulting in a knock-out mutation. In contrast, making a specific, template-directed mutation at a DSB requires homology-directed repair (HDR), which is active during the S-G2 phases of the cell cycle [
6,
7,
8]. Since NHEJ predominates over HDR in most cells [
5], knock-in editing is not very efficient, and, therefore, different strategies have been developed to improve HDR-mediated gene editing (reviewed in [
9]).
2. Strategies to Increase HDR-Dependent CRISPR-Cas9 Mediated Genome Editing
2.1. Inhibiting NHEJ/Promoting HDR Globally to Increase CRISPR Knock-In Editing
Several of the approaches to increasing HDR-dependent gene editing involve shifting the balance between HDR and NHEJ (reviewed in [
10,
11,
12]). Inhibiting NHEJ or promoting HDR, either genetically or pharmacologically, leads to increases in knock-in efficiency [
13,
14,
15,
16,
17,
18] (
Figure 1, middle panel). Likewise, restricting Cas9 activity or expression to the S-G2 phases of the cell cycle, when HDR is active, also improves the knock-in yield (a comprehensive review of this topic can be found in [
19]). Some of these methods include synchronizing the cells by treating them with drugs such as nocodazole (inhibitor of microtubule polymerization) [
20] or the cyclin-dependent kinase inhibitor indirubin [
21]. Another efficient method to restrict Cas9 activity to the S-G2 phases of the cell cycle is by fusing Cas9 to a fragment of a cell cycle-regulated protein, such as geminin [
22]. Geminin is targeted for proteasomal degradation by the cell cycle-regulated APC-Cdh1, resulting in low levels in the G1 phase of the cell cycle and high levels during S/G2/M.
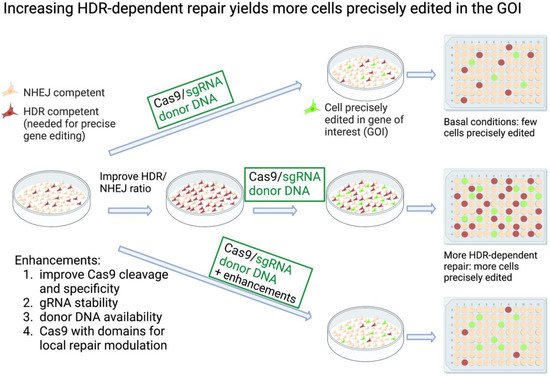
Figure 1. Increasing HDR-dependent repair improves yield of CRISPR knock-in cells. Knock-in HDR-dependent mutagenesis at a gene of interest (GOI). At the top, the basal conditions are shown, where few of the cells are HDR-competent. A fraction of these can be edited at the GOI following transfection with Cas9/gRNA and donor DNA. Isolating the desired knock-in edited cells from the total population can be challenging. In the center, cells are treated either genetically or pharmacologically to alter the HDR/NHEJ ratio. As a result, a higher yield of knock-in edited cells is achieved. The bottom row illustrates some methods used to improve HDR-dependent CRISPR editing. The increases in precisely edited cells depend on the method, the target cells and GOI. Created with BioRender.com.
2.2. Improving Cas9 Cleavage and Specificity
The Cas9 endonuclease most commonly used in genome engineering is derived from
Streptococcus pyogenes (SpCas9). While it is efficient and easy to use, Cas9 from other sources or Cas9 which has been improved through rational design or directed evolution can offer advantages (reviewed in [
12] (
Figure 1, lower panel). For example, newer high-fidelity versions of Cas9 [
23,
24,
25,
26] reduce the possibility of off-target cutting. SpCas9 generates a blunt-ended DSB [
2], which, as mentioned above, is predominantly processed by NHEJ, making indels. To produce a staggered cut that lends itself to precise editing via NHEJ [
27], alternate programmable nucleases such as Cpf1 can be utilized [
28]. Another way to improve specificity and produce a staggered cut is to use a dimeric RNA-guided FokI nuclease (RFN) [
29] or fCas9 [
30] comprised of a pair of catalytically dead Cas9, dCas9, fused to FokI. By using pairs of gRNAs to target the desired locus, the two FokI domains cleave the DNA according to the programmed spacing, producing a staggered cut. Because two gRNAs are required for targeting, specificity is increased.
2.3. Modifications of the Guide RNA and Donor DNA to Improve Editing Efficiency
Another way to improve editing efficiency is to use modified gRNA (for a comprehensive review of this topic, see [
31]) (
Figure 1, lower panel). Chemically modified gRNA can serve to stabilize the gRNA by inhibiting its degradation and can improve the association of the tracrRNA with the crRNA [
32,
33]. Modification of the gRNA can also be used to improve HDR by bringing the donor DNA in close proximity to the cut site. In a method conceptually similar to the pegRNA described below, Lee et al. designed a unique RNA–DNA hybrid, where the RNA comprises the sgRNA and the DNA serves as donor [
34]. The use of this construct increases HDR editing three-fold. In this work, they also demonstrated that HDR is improved two-fold by using a fluorescently labeled donor DNA and enriching the cells that took up the donor DNA by FACS sorting. The Marson lab has developed improved methods for the efficient editing of primary human T cells using non-viral genome targeting, finding ssDNA templates preferable to linear dsDNA, due to reduced toxicity and the chance of random integration [
35,
36]. A hybrid ssDNA template with dsDNA ends used for recruiting Cas9 further increased the yield of correctly modified cells, with knock-in efficiencies of up to 62%.
2.4. Promoting HDR Specifically at the Break Site
Recruiting HDR improving factors to the break site is another way to increase the yield of knock-in mutations (
Figure 1, lower panel). HDR effectors such as CtIP, Rad52, or Mre11 fused to Cas9 promote HDR two-fold [
37]. Another strategy implemented by Tran et al. is to use CtIP fused to the MS2 phage coat protein. This enables recruitment of the CtIP-MS2 to the cut site via MS2 binding sites on an extension of the gRNA. Since Cas9 is a rather large protein, approximately 160 kDa, fusing large domains to it can limit expression options, such as vectors with a limited payload. Using smaller functional domains, or recruiting strategies such as the MS2 loops, can avoid these issues. Fusion of the CtIP N-terminal 296 aa fragment to Cas9, for recruitment of HDR effectors, improves HDR-dependent editing by two-fold or more [
38]. Two-fold improvement in HDR editing was also achieved with Cas9 fused to a 126 aa recruiting domain for the MRN complex (Mre11/Rad50/Nbs1) that is responsible for DNA resection needed for HDR. In this instance, the domain is derived from the HSV-1-encoded protein UL12 [
39]. Alternatively, the fusion of Cas9 to a 413 aa dominant-negative 53BP1 fragment, DN1S, [
40] serves to inhibit NHEJ locally at the targeted break site, which avoids potential random mutations caused by global inhibition of NHEJ. Several of these strategies were compared for their ability to edit the
HBB (hemoglobin subunit beta) locus in human hematopoietic stem and progenitor cells (HSPCs) [
41], confirming that the use of these modified Cas9 constructs can improve HDR-dependent editing in this system by two-fold. The
HBB gene is mutated in sickle cell anemia and beta-thalassemia; thus, finding potent methods to effectively repair the mutation in patient-derived stem cells would have clear clinical benefits. Although doubling the yield of correctly edited cells may suffice for certain targets and cell lines, for most systems, more work needs to be conducted to optimize the overall yield of the correctly edited cells, especially regarding clinical applications.
2.5. Editing without a DSB—Circumventing the HDR Requirement
The repair of a DSB often generates undesired complications, as discussed above, so alternative strategies have been developed where the DSB is avoided altogether. These methods allow certain knock-in mutations to be made without the need for HDR. Base editing is a method using a catalytically inactive Cas9 fused to a base editor to accomplish specific base substitutions at the targeted site [
42]. This method is efficient, with 15–75% editing success, but is limited to point mutations. Prime editing (PE) enables the insertion of longer fragments through the use of a catalytically impaired (nickase) Cas9 fused to a reverse transcriptase [
43]. The donor template sequence is fused to the end of the gRNA (prime editing guide RNA, pegRNA), and the reverse transcriptase creates the donor DNA locally at the targeted site. As with base editing, this strategy avoids many of the pitfalls associated with the repair of DSBs and has an efficiency comparable to the HDR-dependent editing [
43]. However, this method too is restricted to shorter insertions that can be encoded by the pegRNA. To improve upon this method, enabling all the components to be delivered by a single vector, Wolff et al. developed piggyPrime, a transfected single vector system based on piggyBac DNA transposition for genomic integration of all prime editing components [
44]. In this system, the editing components (Cas9, etc.) remain integrated in the genome; thus, this system is not scarless. Eggenschwiler et al. presented a similar system, but with the ability to excise the prime editing components following the editing step using the piggyBac system for scarless editing [
45].
This entry is adapted from the peer-reviewed paper 10.3390/ijms231911919