Diabetic foot ulcer (DFU) is one of the major complications of diabetes. Wound healing under diabetic conditions is often impaired. This is in part due to the excessive oxidative stress, prolonged inflammation, immune cell dysfunction, decreased infection control, delayed re-epithelialization, and decreased angiogenesis present at the wound site. Heme oxygenase-1 (HO-1) is the rate-limiting enzyme in heme degradation generating carbon monoxide (CO), biliverdin (BV) which is converted into bilirubin (BR), and iron. HO-1 is a potent antioxidant. HO-1 can act as an anti-inflammatory, proliferative, angiogenic and cytoprotective enzyme, in wound healing suggesting that HO-1 modulation could be a potential therapeutic approach for the treatment of diabetic foot ulcers.
1. Heme Oxygenase-1
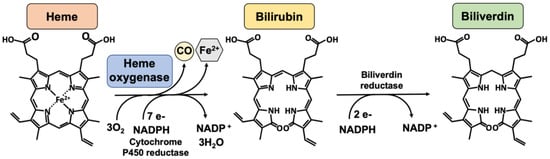
Heme oxygenases consist of a family of enzymes responsible for heme (a pro-oxidant agent) catabolism, in a reaction that produces carbon monoxide, biliverdin, which is consequently reduced to bilirubin by biliverdin reductase, and iron (
Figure 1) [
18]. Two HO isoforms are responsible for this catalytic activity, an inducible enzyme, HO-1, and a constitutively active enzyme, HO-2. HO-3 is not well known and does not have enzymatic activity [
51].
Figure 1. The heme oxygenase enzymatic reaction. Heme oxygenases degrade heme to sequentially generate carbon monoxide (CO), ferrous iron (Fe2+), and biliverdin. The reaction requires 3 mol of molecular oxygen and 7 electrons from NADPH-cytochrome P450 reductase. Bilirubin is subsequently reduced to bilirubin by an NADPH-dependent biliverdin reductase. Legend: NADPH, nicotinamide adenine dinucleotide phosphate.
HO-1 is an ubiquitous stress protein, with a 32-kDa molecular weight, expressed in low quantities under normal conditions except in tissues that involve the degradation of senescent red blood cells, such as the spleen, liver, and bone marrow [
51]. The expression of HO-1 can be induced in response to either exogenous or endogenous stimuli, including ultraviolet (UV) irradiation, natural phytochemicals, statins, heavy metals, heat shock, inflammatory stimuli, heme, oxidative stress, cobalt protoporphyrin-IX (CoPP), iron starvation, hyperoxia, and hypoxia [
19,
20,
21,
52]. Oxidative stress can activate signaling pathways such as the mitogen-activated protein kinases (MAPK), protein kinase C (PKC), 5′-AMP-activated protein kinase (AMPK), and phosphoinositide 3-kinase/protein kinase B (PI3K/Akt), which induce HO-1 expression through the transcription factor nuclear factor erythroid 2–related factor 2 (Nrf2) [
53,
54]. Nrf2 disassociates from its inhibitor, kelch-like erythroid cell–derived protein 1 (Keap1), translocating to the nucleus where it induces the expression of several key antioxidant genes including HO-1 [
55]. Other transcription factors are known to induce HO-1 expression, including activator protein-1 (AP-1), signal transducer and activator of transcription 3 (STAT-3), Yin Yang 1 (YY1), and hypoxia inducible factor (HIF)-1 alpha, through stimuli such as oxidative stress, hypoxia, heme, and IL-6 [
53,
54]. On the other hand, BTB and CNC homology 1 (Bach)1 is a transcription factor which inhibits HO-1 expression [
24]. Moreover, HO-1 is inhibited by metalloporphyrins, including tin protoporphyrin-IX (SnPPIX) and zinc protoporphyrin-IX (ZnPPIX), which compete with heme for the HO-1 binding site [
56,
57]. The metalloporphyrins, based in the porphyrin structure, have been tested for their ability to competitively inhibit the degradation of the heme group. Both SnPPIX and ZnPPIX have been shown to strongly inhibit heme degradation. However, SnPPIX was found to be the most potent inhibitor of HO activity [
58].
HO-1 is known to be localized in the endoplasmic reticulum, anchored via the COOH terminus. In the endoplasmic reticulum, HO-1 is in the proximity of cytochrome P450 reductase, which is required for its high enzymatic activity [
59,
60]. Furthermore, HO-1 has been observed in other cellular localizations such as in caveolae, mitochondria, and in nucleus, which has been associated with the truncation of the COOH terminus and loss of enzymatic activity, suggesting that HO-1 may have other bioactive actions other than catalyzing heme degradation [
61,
62].
The effects of HO-1 in tissue protection are evident in patients who have HO-1 genetic deficiencies, with accumulation of heme and iron, leading to increasing inflammation and ROS. This in turn results in damage of the liver, kidney, and vasculature [
63]. Moreover, mice with HO-1 deficiency present high levels of heme in circulation and concomitant increases in inflammation [
64]. In addition, in several animal models of disease, overexpression of HO-1 levels has been shown to confer protection to the heart, lung, and vasculature, as well as against skin injury [
34,
65,
66].
2. HO-1 in Wound Healing
Immediately after injury, HO-1 is induced in the wounded tissues [
22,
23]. During hemolysis, the pro-oxidant heme is released, triggering inflammation and oxidative stress [
90]. Heme is a strong inducer of HO-1, which degrades heme and has an important protective role against the oxidative and inflammatory results in the local wounded tissue [
22,
90]. It is known that the effect of preventing heme toxicity by HO-1 is mediated by the activation of Nrf2 [
75].
The importance of HO-1 in wound healing has been confirmed not only by using HO-1 deficient mice but also by pharmacological inhibition of HO-1, which results in a significantly delayed wound closure [
34,
90,
91]. In both models, the wounds showed an increase in oxidative stress and inflammation, as well as impaired wound re-epithelialization and angiogenesis [
34,
90,
91]. In addition, hemin, an inducer of HO-1, was able to significantly improve wound healing, acting as an anti-inflammatory agent [
92].
HO-1 is also a key player in the regulation of diabetic wound healing, but the effect is limited due to the high basal levels of oxidative stress in diabetes that are responsible for the damage of proteins, lipids, and DNA, which can lead to tissue impairment. Under this condition, the increased oxidative stress and inflammation that further occurs after wounding can result in impaired wound healing. Diabetic skin tissue collected from peri-wound regions showed higher levels of oxidative stress markers than peri-wound skin regions from non-diabetic patients. Similarly, it has been shown that HO-1 levels are increased in rodent diabetic wounded tissues via Nrf2 activation. This may suggest a compensatory mechanism for the increase in oxidative stress, demonstrating the important contribution of HO-1 in diabetic wound healing [
93].
Animal models of diabetes have also been used to better understand the role of HO-1 in diabetic wound healing. HO-1 levels are increased after wounding in non-diabetic and db/db mice, a type 2 diabetic animal model, particularly in the initial wounding phase [
34]. However, the increase in HO-1 levels in diabetic wounds appears to be delayed, which may trigger the impaired wound process overserved under these conditions. Similarly, the induction of HO-1, using an intradermal injection of hemin, was impaired in diabetic mice, showing a decrease in HO-1 levels [
34]. On the other hand, the local delivery of HO-1 transgene by adenoviral vectors improved wound healing in diabetic db/db mice by increasing neovascularization [
34]. Furthermore, the HO-1 inducer hemin has been shown to promote wound healing in diabetic rats by reducing inflammatory cytokines, increasing antioxidant defenses, and promoting angiogenesis [
26,
76]. Hemin has been shown to decrease oxidative stress by an increase in the antioxidant defenses in several experimental models [
94,
95], and to decrease inflammatory cytokines in animal models of disease [
94,
96]. However, since the use of the HO-1 inhibitor SnPPIX, in the presence of hemin, has shown the opposite effect of hemin, delaying wound healing in diabetic rats, this suggests that the effects of hemin may be through the induction of HO-1 [
26,
76].
3. Therapeutic Potential of HO-1 Induction for DFU Treatment
Several HO-1 inducing agents include antioxidant compounds, mostly plant-based, found in the diet. These compounds activate the Nrf2 system that enhances the expression of several cytoprotective proteins such as HO-1. Among these natural compounds are resveratrol [
128], curcumin [
129], quercetin [
130], epigallocatechin gallate [
131], sulforaphane [
132], and others. The efficacy of these dietary compounds at promoting wound healing when used in in vitro and in vivo models, through the induction of several protective proteins including HO-1, has led to the hypothesis that these may be used as pharmaceuticals for DFU.
Pharmaceutical compounds, such as dimethyl fumarate (DMF), have been found to activate Nrf2 [
133]. DMF has shown therapeutic effects for multiple sclerosis and has been approved for clinical use [
134,
135]. DMF was shown to improve wound healing in diabetic mice [
136], and since it is used in the clinic, it could be repurposed as a treatment for DFU. Since Nrf2 regulates multiple effector enzymes, the effects of Nrf2-inducing compounds, such as DMF, are not necessarily specific to HO-1. Further research is needed to develop safe and effective Nrf2 activator compounds for clinical use.
Hemin, a known inducer of HO-1, has also been approved for acute intermittent porphyria treatment [
137]. Hemin was shown to improve diabetic wound healing in animal models. Thus, hemin may also be a potential drug candidate for DFU treatment.
Additionally, other pharmacological options include the use of BV/BR based therapies, which have proven to be effective in promoting wound healing in animal models [
25,
103,
110] and/or the direct administration of CO via inhalation or CO-releasing molecules [
24,
29,
52,
108,
112,
113]. The exogenous application of these substances as pharmaceuticals, even at low concentrations, may not necessarily have a similar effect as the endogenous product; thus, further research is needed to evaluate their potential therapeutic effect.
This entry is adapted from the peer-reviewed paper 10.3390/ijms231912043