Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
In adipose triglyceride (AT), but also in other organs, three major enzymes are involved in lipolysis—adipose triglyceride lipase (ATGL), hormone-sensitive lipase (HSL), and monoacylglyceride lipase (MGL). TGs, specifically triolein, are very often used as a substrate to measure HSL and ATGL activity despite the fact that HSL hydrolase activity is up to 10-fold higher for diglycerides (DGs) compared to TGs. The activity of lipolysis is finely regulated by multiple signals, with catecholamines, insulin, growth hormone, and natriuretic peptides being the main hormonal regulators.
- adipose triglyceride lipase
- hormone-sensitive lipase
- acetyl-CoA carboxylase
1. Adipose Triglyceride Lipase (EC 3.1.1.3)
ATGL (gene code PNPLA2; Patatin-Like Phospholipase Domain Containing 2), also called desnutrin, is a 54 kDa serine hydrolase [1] (catalytic dyad Ser, Asp [2]), discovered in AT by Zimmerman et al. in 2004 [3]. It is expressed not only in AT but also in the liver, testis, skeletal and cardiac muscle, intestine, and β-cells [4][5][6]. It is attached to the lipid droplet by a hydrophobic stretch of amino acids 315–360 [2] and performs the first step in TGs hydrolysis there. ATGL has also been reported to have transacylase and phospholipase activities that appeared to be lower than its TG hydrolase activity [1][4]. ATGL has two phosphorylation sites localized in the C-terminal region of the enzyme (Ser404 and Ser428). In mouse AT, phosphorylation of these serine residues is carried out by AMP-activated protein kinase (AMPK) and leads to an increase of ATGL activity. In humans, it is not yet fully understood whether ATGL is phosphorylated by AMPK or Protein Kinase A (PKA) [7].
Important regulation of ATGL activity in a cell is provided by a coactivator protein annotated as α/β-fold domain-containing protein 5 (ABHD5), also known as comparative Gene Identification 58 (CGI-58) [8], in coordination with a battery of proteins including the lipid droplet coating proteins from perilipin (PLIN) family. Phosphorylation of PLIN1, which is the major lipid coating protein in adipocytes, is required for the release of ABHD5 from his docking site. ABHD5 then binds to and activates ATGL [9]. PLIN5, which is expressed predominantly in skeletal muscle, binds to both ABHD5 and ATGL itself and activates it independently of PLIN1 [10]. In mice, PLIN2 is known to affect lipolysis only modestly and is not activated by PKA. Overexpression of PLIN2 decreases the access of ATGL onto the lipid droplet and thereby suppresses lipolysis [11]. Data on the role of PLIN3 and 4 in the regulation of ATGL activity are missing. Another coactivator of ATGL is a pigment epithelium-derived factor (PEDF), called by newer nomenclature as serpin F1. The binding of this monomeric 50-kDa protein to ATGL induces TGs hydrolysis [9]. On the other hand, AGTL activity can be inhibited by the protein encoded by G0/G1 switch gene 2 (G0S2) [12] as well as by metabolites, particularly by long-chain acyl-CoA [13].
In addition to biological negative regulators, there are three synthetic molecules that inhibit ATGL activity. One of the first synthetic ATGL inhibitors is Atglistatin, the derivative of urea, which is a selective, competitive inhibitor of ATGL [14]. It is active only in mouse tissues (IC50 0.7 µM) and fails to inhibit more than 10% of ATGL hydrolase activity in human AT [14][15]. Atglistatin affects the patatin-like domain. Sequences 23–101 and 146 amino acids determine the ATGL binding region [16]. Another ATGL inhibitor is Orlistat (tetrahydrolipstatin, ester of L-leucine, and hydroxy-FA with a β-lactone ring). Orlistat is however not a specific inhibitor of ATGL as it inhibits effectively also MGL, HSL, diacylglycerol lipase (a key enzyme in the biosynthesis of the endocannabinoid 2-arachidonoylglycerol [17]), carboxylesterase 1 and even FA synthase (FAS) [18]. Moreover, its major therapeutic potential relates to the inhibition of gastric and pancreatic lipases [19]. Orlistat reduces dietary fat absorption and thus it is currently used as an anti-obesity drug [20][21]. Cay10499 (derivative of methylphenyl ester of N-derived carbamic acid) inhibits ATGL by 95% in humans (IC50 66 nM), but it also inhibits HSL, MGL, diacylglycerol lipase, ABHD6 and, carboxylesterase 1 very efficiently (60–95%) [15]. Recently, a specific inhibitor of human ATGL called NG-497 has been developed. It is a selective, competitiv reversible inhibitor with an IC50 of 1.0 µM that binds to human ATGL in a patatin-like domain in the amino acid sequence 60–146. Although this domain is highly conserved between species, even minor differences in amino acid sequence in pigs, primates, and humans vs. other mammals (dog, goat, mouse, rat) apparently substantially affect NG-497 vs Atglistatin efficacy, as shown by Grabner et al. [16]. Thus, species-specific sequence differences (between 60 and 146 amino acids) and the associated structure of the active domain of ATGL are likely to be crucial for the selectivity and efficacy of the inhibitors and, hypothetically, may also serve to regulate ATGL in vivo, which have not yet been revealed. The structure of ATGL inhibitors is shown in Figure 1 and information on them is summarized in Table 1.
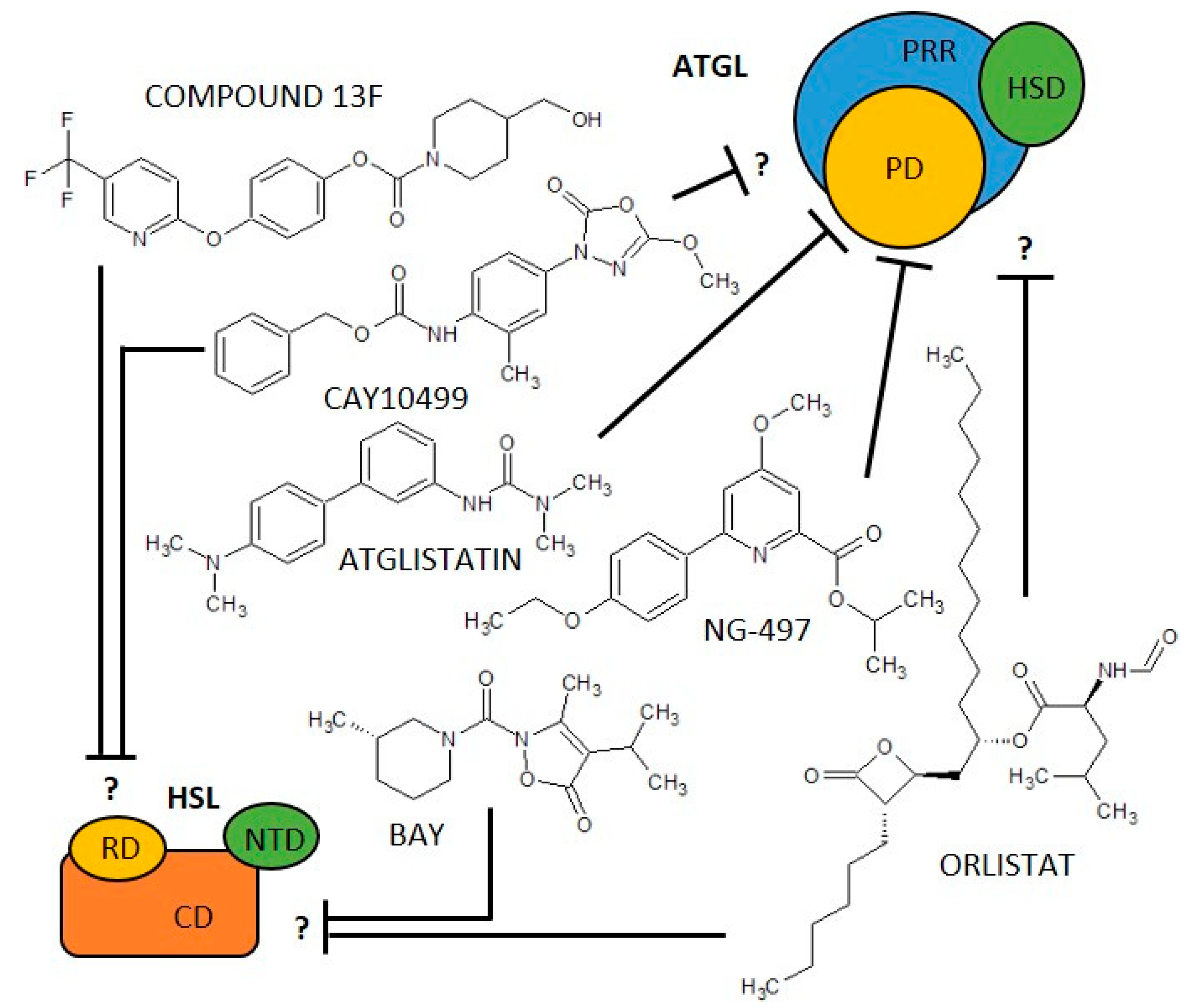
Figure 1. Scheme of adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL) structure/domains and their inhibitors. ATLG domains: PD, Patatin-like Domain; PRR, Patatin Related Domain; HSD, Hydrophobic Stretch Domain. HSL domains: CD, Catalytic Domain; NTD, N-Terminal Domain; RD, Regulatory Domain. ? = it is not described which domain is affected.
Table 1. An overview of (co)activators and inhibitors of selected lipolytic and lipogenic enzymes.
| Enzyme (EC Code) |
(Co)Activators | Negative Regulators | (Semi)Synthetic Inhibitors | ||||
|---|---|---|---|---|---|---|---|
| Name(s) | Organism or Cell Line |
Enzyme Specificity 1 | Inhibition Type |
IC50 [nM] | |||
| ATGL (EC 3.1.1.3) |
ABHD5 [8][9][10][22][23] PEDF [9][24][25] PLIN1 [9][10][26] PLIN5 [10][27] |
G0S2 [12][23][28] long acyl-CoA [13] |
Atglistatin [14][15][16] |
dog, goat, marmoset, mouse, rat | yes | competitive, reversible [14] |
700 [14] |
| CAY10499 [15][29] |
human, mouse | no | unknown | 66 [15] | |||
| NG-497 [16] | human, rhesus monkey | yes | competitive, reversible [16] |
1300 [16] | |||
| Orlistat [15][30][31] |
human, mouse | no | irreversible [31] | 1.2 [15] | |||
| HSL (EC 3.1.1.79) |
FABP4 [32][33] PKA [34][35] PLIN1 [9][34] |
AMPK [36][37] PLIN2 [38] |
BAY 59-9435 [39][40] |
3T3-L1, human, mouse, rat | yes | non-competitive, reversible [39] | 5 [39] |
| CAY10499 [15][41] |
human, mouse, rat | no | unknown | 79.8 [15] | |||
| compound 13f [15][42] |
human, mouse | no | reversible [42] | 110 [42] | |||
| Orlistat [15][43] |
human, mouse, rat | no | irreversible [31] | 4230 [43] | |||
| ACC (EC 6.4.1.2) |
citrate [44][45] glutamate [46][47][48] |
AMPK [49][50] HS-CoA [51] malonyl-CoA [51] long acyl-CoA [51][52] |
CP-640186 [53][54][55][56][57][58][59] |
human, monkey, mouse, rat | yes | non-competitive, reversible [54] | 53–61 [53] |
| ND-630, ND-646 [54][58][60][61][62] |
human, rat | yes | reversible [60] | 1.7–6.1 [60][62][63] | |||
| PF-05175157 [64][65][66] |
dog, human, rat | yes | unknown | 27–33 [64] | |||
| Soraphen A [54][55][56][57][59][63] |
HepG2, LNCaP, PC-3M | yes | unknown | 1–5 [63] | |||
| TOFA [53][54][58][64][65][66][67][68][69][70] |
human breast cancer cells, human colon carcinoma cells, human lung cancer cells | no | allosteric, irreversible [67] | 51 [70] | |||
| FAS (EC 2.3.1.85) |
unknown | unknown | cerulenin [69][71][72][73][74][75][76] |
human, mouse, MCF-7 breast cancer cells, MDA-MB-231, SKBr-3 | yes | non-competitive, irreversible [75] | 70,000–79,000 [74] |
| C75 [72][77][78][79] |
HL60 cells, human, mouse, SKBr-3, rat | yes | competitive, irreversible [78] | 26,380 [79] | |||
| Orlistat [18][80][81][82] |
PC-3, human, mouse | no | irreversible [31] | 900 [82] | |||
| Triclosan [83][84][85] |
HepG2, MCF-7, SKBr-3 cells | no | reversible [83] | 6900–50,000 [83][86] |
|||
1 Enzyme specificity—yes—the inhibitor is specific to a given enzyme; no—the inhibitor acts on several different enzymes or has several non-specific effects.
2. Hormone-Sensitive Lipase (EC 3.1.1.79)
HSL (gene code LIPE; Lipase E) is—depending on the isoform—84 to 130 kDa [1][34] serine hydrolase, expressed in AT and adrenal glands, with lower expression in cardiac and skeletal muscle and macrophages [1]. HSL consists of three domains. The C-terminal domain contains the active site including the catalytic triad (Ser, Asp, His) [34], while the N-terminal domain interacts with FA binding protein-4 (FABP4). The third domain represents the regulatory stretch [2][87]. HSL performs mainly the second step of TG hydrolysis—it hydrolyses DGs generating MGs and free FA. Besides, HSL hydrolyses also TGs, cholesteryl esters, MGs, and retinyl esters [1][2][34].
The most important activators of HSL are catecholamines, which act through adrenergic receptors and increase the level of cAMP in the cell, followed by activation of PKA [34]. The third domain of human HSL contains serine residues (Ser552, Ser554, Ser589, Ser649, and Ser650) that are crucial for the modulation of enzyme activity through their phosphorylation. Phosphorylation of fof these serine residues enhances HSL activity, while phosphorylation of Ser554 by AMP-activated kinase inhibits HSL activity [87]. The phosphorylation of HSL and also HSL coactivator—PLIN1—by PKA is necessary for the translocation of HSL from cytosol to the lipid droplet and the initiation of HSL-mediated lipolysis [9][34]. On the other hand, HSL activity may be inhibited upon binding of PLIN2 as shown in cardiomyocytes [38]. As mentioned above, the N-terminal domain of HSL interacts with FABP4, a cytosolic lipid-binding protein expressed in adipocytes, which facilitates FA uptake. The interaction of HSL with FABP4 appears to increase HSL activity after PKA activation [32].
Similar to ATGL, inhibition of HSL has been suggested as a pharmacological approach to reduce free FA levels and improve peripheral insulin resistance. Accordingly, in the last 20 years, various HSL inhibitors, including natural and synthetic products, have been described for the treatment of diabetes and lipid disorders [39]. Moreover, the use of inhibitors and activators of HSL activity is indispensable in basic research for deciphering the HSL’s role in various pathophysiological conditions [88][89][90][91]. The only specific, reversible non-competitive inhibitor of HSL is BAY 59-9435 (BAY), a derivative of 5-(2H)-isoxazolonyl urea. BAY is able to inhibit human, mouse, and/or rat HSL. Nevertheless, while mouse HSL can be inhibited by 90% by BAY, the inhibition of human HSL reaches only 30% [39]. Further, there are several non-specific HSL inhibitors that partially block the activity of HSL, while blocking other lipases as well. Among them is Compound 13f, an ester of 4-hydroxymethyl-piperidine-1-carboxylic acid, which reversibly inhibits human HSL from 17% (5 µM) [15][42] while also acting on other lipases such as carboxylesterase 1, MGL, or ABHD6 [15]. Cay10499 (derivative of methylphenyl ester of N-derived carbamic acid) inhibits mouse and human HSL by 95%, and 67%, respectively, but it also inhibits ATGL, MGL, diacylglycerol lipase, ABHD6, and carboxylesterase 1 very efficiently (60–95%) [15]. Thus, Cay10499 can be successfully used to block all major members of TAG lipolysis, hydrolysis of endocannabinoids, and cholesterol esters [15]. Orlistat also irreversibly blocks, in addition to ATGL, mouse, human, and rat HSL function [15][31][43]. The structure of HSL inhibitors is shown in Figure 1, and information on them is summarized in Table 1.
3. Assays for Measurement of ATGL and HSL Activity
As mentioned above, ATGL and HSL activity play important role in the regulation of metabolism and pathophysiology of a number of diseases [39][92]. Therefore, the measurement of the activity of these enzymes is widely applied in metabolic research [93][94][95][96].
As the total lipolytic enzyme activity is most conveniently measured at the product level, i.e., glycerol and free FA, the activity of individual lipases can only be analyzed when using purified/isolated enzymes. The only other possibility is to effectively inhibit the activities of all other lipases or to use an enzyme-specific reaction, if one exists. For example, to measure the activity of ATGL, the lipase activity of HSL and other lipases must be inhibited [15][97]. A scheme of inhibitor action on lipolytic enzymes is shown in Figure 2. It is also possible to focus on and analyze one of the esterase activities that none of the other lipases exhibit.
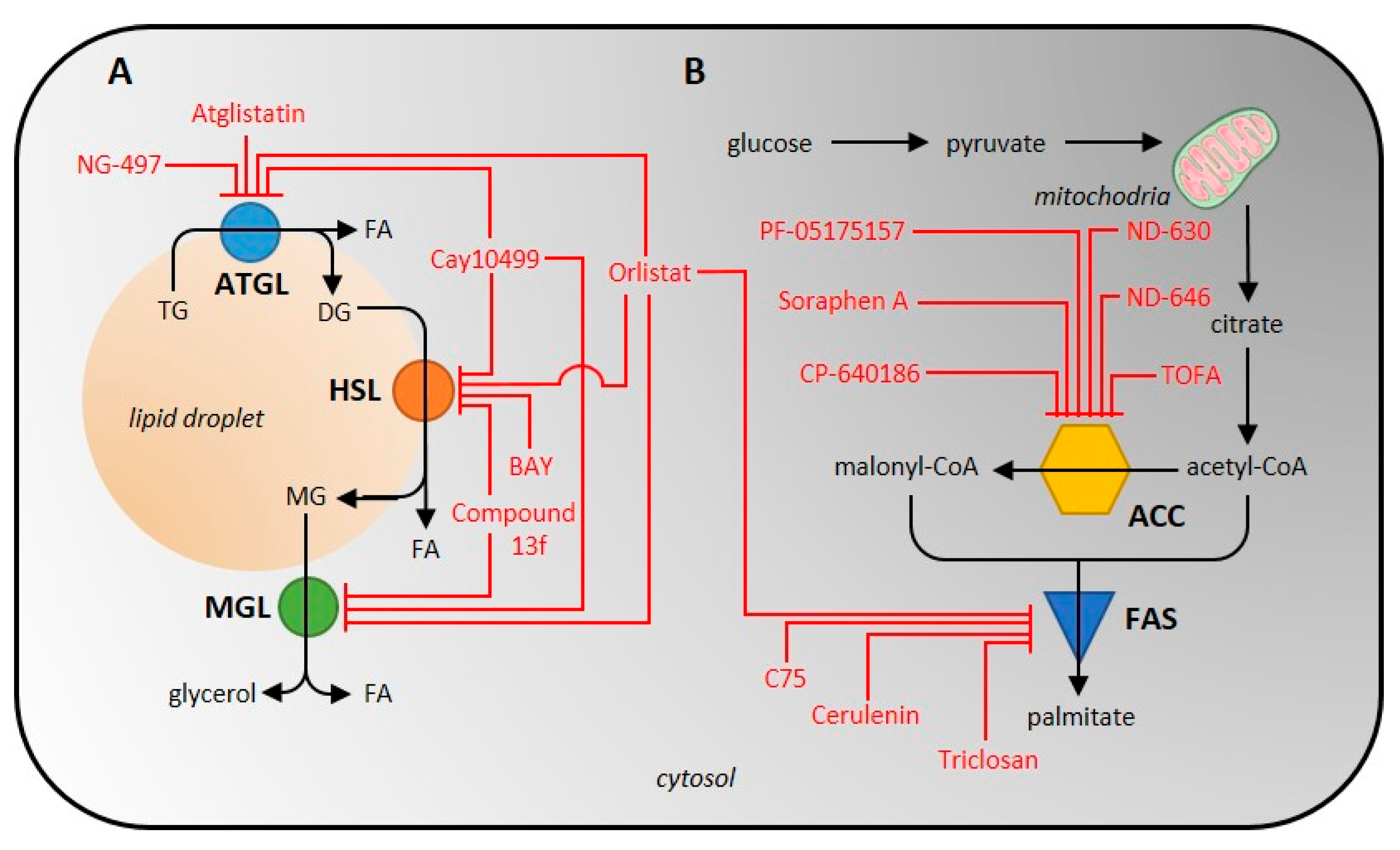
Figure 2. Scheme of inhibitor action on individual enzymes of the lipolytic (A) and de novo lipogenic (B) pathway. ATGL, adipose tissue triglyceride lipase; HSL, hormone-sensitive lipase; ACC, acetyl-CoA carboxylase; FAS, fatty-acid synthase.
3.1. Isolation and Purification of the Lipolytic Enzymes
Isolation of lipases is carried out by disrupting cells or tissue with a Potter-Elehjem homogenizer, freezing, or sonication [97][98][99][100][101][102][103][104][105][106][107][108][109]. This is followed by centrifugation, the conditions of which can vary considerably across sample types, i.e., 1000–110,000× g, 1–90 min, 4 °C [97][98][99][100][101][102][103][104][105][106][107][109][110][111][112][113]. After homogenization, recombinant or tissue-derived lipase can be purified by Q-sepharose chromatography, phenyl-sepharose chromatography, QAE-sephadex chromatography [98][102], hydroxyapatite chromatography, or gel filtration chromatography with triacylglycerol-containing acrylamide and agarose (Ultrogel AcA 34) [102].
Another approach to isolating lipolytic enzymes is to isolate lipid droplets that contain various lipases and their co-activators [114]. Isolation of lipid droplets has been successfully performed across many cell types, even on some tissues such as rat and mouse liver, bovine and mouse mammary glands, and others [115]. To homogenize the sample, a homogenizer can be used, or high pressures can be applied using nitrogen bombs. After gentle spinning, the supernatant is removed and ultracentrifuged at 10,000–182,000× g, 30–60 min, 4 °C [115][116]. Ultracentrifugation with a density gradient of 5% sucrose can also be used [116]. It should be kept in mind that the higher the speed during centrifugation or its duration, the higher the risk for the destruction of lipid droplets and also removal of proteins from lipid droplets.
3.2. Conditions Affecting the Ex Vivo Activity of ATGL and HSL
ATGL and HSL are neutral lipase with a pH optimum of 7.0 [97][98]. Remarkably, when the pH is changed by ±1, the activity of ATGL decreases by almost 50% [97]. The activity of ATGL/HSL can also be inhibited by the detergents used; therefore, it is necessary to dilute the lysate appropriately to avoid detergent interference [97][98][102]. Coactivators can be used to increase enzyme activity. ATGL activity increases 2–20-fold in the presence of coactivator ABHD5 [8][97][100], and is highest when is ABHD5 is emulsified with phosphatidylcholine/phosphatidylinositol (PC/PI) [97].
It should be noted that the isolated/purified enzyme may not have its activators and other regulatory molecules available, so the measured activity is unlikely to reflect the actual in vivo activity. In order to achieve the most physiological situation, it would be advisable to have the activators, metabolites, and ions present in the reaction mixture, preferably in physiological concentrations.
3.3. Radioisotope Methods for Measurement of Lipolytic Enzyme Activity Determination
In fact, a radioisotope assay with a radiolabeled [9,10-3H]-triolein emulsified with a PC/PI mixture [5][99][100][101][102][103][107][108][109][110][111][117] or dissolved in toluene [97][112] is one of the earliest methods used to measure ATGL or HSL activity. The method is based on the hydrolysis of triolein to free [9,10-3H]-oleate and analysis of this product by liquid scintillation counting [5][97][99][100][101][102][103][106][107][108][109][110][111][112][117]. The higher specificity of HSL towards DG may influence the resulting activity. In unpurified lysates, it is necessary to inhibit lipases outside of the researchers' interest, as discussed above.
HSL activity can also be measured by substrate specificity to cholesterol esters and DGs (for example, neither MGL nor ATGL have cholesteryl esterase activity [1]). In the first case, the radiolabeled substrate cholesteryl-[1-14C]-oleate is hydrolyzed to free cholesterol and [1-14C]-oleate [106][109][110][112][113]. In the second case, a radiolabeled [3H]-oleoylmonoalkylglycerol, such as [3H]-oleoyl-2-O-oleylglycerol, is hydrolyzed to monoalkylglycerol and [3H]-oleate [97][98][105]. This substrate has several advantages: firstly, DG is the preferred lipid substrate for HSL, and secondly, monoacylmonoalkylglycerol does not form a substrate for MGL and therefore MGL activity does not interfere in this assay [98].
Generally, in all the mentioned radioisotope methods 0.2–1 mg/mL of total protein is used in the reaction mixture [5][107][109] and the reaction is terminated after 20–60 min of incubation at 25–37 °C [5][97][98][99][100][101][103][104][105][106][107][108][109][110][111][112][113][117] by the addition of a methanol/chloroform/heptane mixture containing 0.1 M potassium carbonate and/or 0.1 M boric acid (pH 10.5) [5][97][100][101][102][103][106][107][108][109][110][111]. After extraction and centrifugation, the radioactivity in the upper phase is determined by liquid scintillation counting [5][97][98][99][100][101][102][103][104][105][106][107][108][109][110][111][112][113][117][118][119].
HSL activity can also be measured using radiolabelled [α-32P]-glycerol. In this assay, glycerol released from acylglycerols (TGs, DGs, or MGs) is phosphorylated by glycerol kinase using [32P]-ATP. After precipitation of the free [γ-32P]-phosphate with ammonium molybdate/triethylamine, the radioactivity of the labeled glycerol in the supernatant is measured by liquid scintillation counting [118][119]. The advantage of this method is the high stability of the measured [α-32P]-glycerol [118]; however, isotope 32P is a β emitter that poses a potential health risk [120]. Since HSL shows esterase activity towards MGs and DGs, the cleavage of MGs to free glycerol by MGL activity needs to be inhibited to increase the specificity of the reaction. Next, another potential bias could be the varying activity of glycerol kinase, which attaches a phosphate group to glycerol.
Radioisotope assays are applicable to the analysis of ATGL and HSL activity in a wide range of samples including recombinant ATGL [97][107][110] or HSL [97][98][110] (produced in monkey embryonic kidney cells COS-7 [97][107], Escherichia coli BJ5183 cells [110], insect Spodoptera frugiperda cells [98], and rat hepatoma cell line McA-RH7777 [110]) and endogenous ATGL and HSL isolated from rat adipocytes [119], 3T3-L1 adipocytes [117], L6 myoblasts, mouse peripheral leukocytes [108][109], human [99][103][104][105][106][112], mouse [100][101], rat [102] and/or porcine [111] AT, mouse liver [5][110][113][117], skeletal muscle, testes [101] or intestine [5] (summarized in Table 2).
Table 2. Summarizing the determination of ATGL, HSL, ACC, and FAS activity measurements.
| Enzyme | Activity Measurement | Biological Material | Reference | ||
|---|---|---|---|---|---|
| Detection Method | Substrate | Detecting Substances |
|||
| ATGL | fluorescence assay |
pyrene-labeled acylglycerols |
pyrene | mouse AT | [121] |
| EnzChek substrate | - | human recombinant protein (HEK 293T cells) mouse recombinant protein (HEK 293T cells) |
[15] | ||
| liquid scintillation counting |
[9,10-3H]-triolein | [3H]-oleate | 3T3-L1 adipocytes, L6 myoblasts | [117] | |
| human AT | [99] | ||||
| human recombinant protein (COS-7 cells) |
[107] | ||||
| McA-RH7777 | [110] | ||||
| mouse gonadal AT | [100] | ||||
| mouse liver | [5][110] | ||||
| mouse peritoneal macrophages | [109] | ||||
| mouse recombinant protein (COS-7 cells) |
[97] | ||||
| peripheral leukocytes | [108] | ||||
| spectrophotometric assay | p-nitrophenyl esters | p-nitrophenol | mouse AT | [121] | |
| HSL | fluorescence assay |
pyrene-labeled acylglycerols |
pyrene | mouse AT | [121] |
| 1-S-arachidonoylthioglycerol | ThioGlo-1 aduct | human and mouse recombinant protein (HEK 293T cells) | [15] | ||
| liquid scintillation counting |
cholesteryl-[1-14C]-oleate | [14C]-oleate | human AT | [106] | |
| McA-RH7777 | [110] | ||||
| mouse liver | [110][113] | ||||
| mouse peritoneal macrophages | [109] | ||||
| [3H]-oleoyl-2-O-oleylglycerol | [3H]-oleate | human recombinant protein (Sf9 cells) rat AT |
[98] [102] |
||
| [32P]-ATP | [α-32P]-glycerol | rat adipocytes | [119] | ||
| [9,10-3H]-triolein | [3H]-oleate | human AT | [103][112] | ||
| mouse AT, skeletal muscle, and testis |
[101] | ||||
| mouse recombinant protein (COS-7 cell) |
[100] | ||||
| mouse gonadal AT | [97] | ||||
| porcine AT | [111] | ||||
| spectrophotometric assay | p-nitrophenyl esters | p-nitrophenol | mouse AT | [121] | |
| ACC | HPLC (reverse phase) |
acetyl-CoA/ malonyl-CoA |
acetyl-CoA/ malonyl-CoA |
mouse 3T3-L1 preadipocytes | [51] |
| liquid scintillation counting |
Na(K)H14CO3 | 1-[14C]-malonyl-CoA | Fao hepatoma cells | [122] | |
| chicken liver | [123] | ||||
| human skeletal muscle | [124] | ||||
| lamb AT | [125] | ||||
| rat AT | [126][127] | ||||
| rat hepatocytes | [128][129] | ||||
| rat liver | [126][130] | ||||
| rat skeletal muscle | [131] | ||||
| spectrophotometric assay | NADH | NADH | chicken liver | [123] | |
| mouse AT | [132] | ||||
| rat liver | [130] | ||||
| NADPH | NADPH | chicken liver | [44] | ||
| FAS | fluorescence assay |
CoA | CPM-CoA adduct | HepG2, human epithelial SKBr-3, rat liver | [133] |
| human lung cancer | [134] | ||||
| GC-MS/LC-MS | [13C]-acetyl-CoA [13C]-malonyl-CoA |
[13C]-palmitate | cow mammary gland | [135] | |
| mouse liver, mouse mammary gland | [76] | ||||
| liquid scintillation counting |
1-[14C]-acetyl-CoA | 1-[14C]-acetyl-CoA | HepG2 | [136] | |
| rabbit mammary gland | [137] | ||||
| rat liver | [138] | ||||
| 2-[14C]-malonyl-CoA | 2-[14C]-malonyl-CoA | BT474 | [139] | ||
| H35-BT, mouse liver, primary hepatocytes |
[140] | ||||
| HepG2 | [136] | ||||
| human liver | [141] | ||||
| LNCaP | [142] | ||||
| pigeon liver | [143] | ||||
| rabbit mammary gland | [137] | ||||
| spectrophotometric assay | NADPH | NADPH | 3T3-F442A | [144] | |
| BT474, MCF-7, MDA-MB-231 breast cancer cells | [145] | ||||
| HCT116, HEK293T | [146] | ||||
| human adipocytes | [147] | ||||
| human AT | [148][149] | ||||
| human liver | [141] | ||||
| lamb AT | [125] | ||||
| mouse AT | [132] | ||||
| rabbit mammary gland | [46][137] | ||||
| rat liver | [77][138] | ||||
| ZR-75-30 | [146] | ||||
3.4. Spectrophotometric and Fluorescence Methods of Lipolytic Enzyme Activity Determination
Other methods for measuring ATGL or HSL activity are based on spectrophotometric [121] or fluorescence [15][121] assays. The spectrophotometric test is performed by the esterase cleavage of p-nitrophenyl esters (acetate, butyrate, or laureate) and the concentration of free p-nitrophenol is measured at 405 nm. In a reaction mixture containing 150 µM p-nitrophenol esters, 5 µg/mL of total protein was used. This method was applied to the mouse white and brown AT [121] (Table 2).
A fluorescence assay uses the ability of the two lipases to cleave pyrene-labeled acylglycerols (Table 2) into free pyrene and acylglycerol, when the fluorescence of the released pyrene is measured. The substrate is dissolved in the PC/PI mixture (3:1, w/w) and 0.25 mg/mL total protein is added to the reaction, which is run for 1 h at 37 °C. The reaction is then terminated with a solution of chloroform:methanol (2:1, v/v) and HCl, followed by an extraction process and separation by TLC chromatography. Fluorescent spots are detected using a CCD camera at an excitation wavelength of 365 nm [121]. Another substrate used to measure lipase activity in fluorescence assay is EnzChek (C58H85BF2N6O6), the commercially developed fluorescent analog of TG. This substrate is successfully cleaved by recombinant human and mouse ATGL or HSL overexpressed in 293T cells (Table 2) and has the advantage of measuring the time dependence of enzyme activity. The reaction mixture contained 2 to 4 mg/mL of total protein. After 30 min preincubation at room temperature, 5 µL of 20 µM EnzChek lipase substrate was added to the reaction mixture, and fluorescence decline was recorded every 30 s for 60–90 min at excitation and emission wavelength at 485 and 510 nm, respectively [15].
HSL activity can be determined also upon the hydrolysis of 1-S-arachidonoylthioglycerol as a substrate. Released 1-thioglycerol spontaneously reacts with ThioGlo-1 to form a fluorescent adduct. The reaction mixture contains 0.11 mg/mL of recombinant HSL and an increase in fluorescence is continuously recorded at excitation and emission wavelength 380 and 510 nm [15].
3.5. Advantages and Disadvantages of the ATGL and HSL Activity Measurement
When choosing a particular radioisotope, spectrophotometric and fluorescence method for measuring the activity of lipolytic enzymes, some specifics should be considered. Liquid scintillation radioassay can be used for a variety of tissues or cell samples, whereas spectrophotometric and fluorescence assay has only been described in tissues where the lipases were overexpressed (293T cells or mouse adipose tissue). Considering the amount of total protein needed for the measurement, the methods are quite similar (0.2–1 mg/mL), with exception of the spectrophotometric assay with p-nitrophenol, which uses only 5µg/mL of total protein. The advantage of fluorescence and spectrophotometric assays is the simplicity of both sample processing and the actual measurement, while the certain disadvantage is the lower sensitivity and lower specificity given by using substrates, which are not primary targets of the enzymes and combined specificity of the enzyme for different substrates—contamination by other enzymes. On the other hand, the spectrophotometric and fluorescence methods have the advantage of measuring kinetics parameters in real-time or end-point mode, while radioisotope methods allow only end-point mode.
A number of isolation procedures have been described for the enrichment of ATGL and HSL in the sample prior to activity assays. After the homogenization of biological material, it is recommended to use chromatographic methods with elution techniques that separate proteins on the basis of size. These methods have the advantage that they can preserve the activity of these enzymes; however, a lysate with proteins of similar molecular weight is obtained. A pure enzyme can be obtained by using antibodies against the enzyme, but this may in turn reduce its activity. In general, however, the higher the number of purification steps, the more the activity decreases. Thus, the compromise between the purity of the enzyme and its activity must be considered.
To increase the specificity of the individual enzymes in these methods, specific substrates or inhibitors suppressing the activity of other lipases are used. Of course, both ATGL and HSL prefer TGs/DGs rather than other esters and it must be taken into account that the activity measured with other substrates will always be slightly lower than if measured with preferred substrates.
Indeed, due to the use of unnatural substrates, the activities measured by the spectrophotometric assay do not match the in vivo activities of the measured enzymes. Radioassay requires a number of additional steps such as termination and extraction, which increases the time and cost burden of the method. Moreover, working with radioactive material requires special approvals, and the detection systems are often very expensive. Fluorescence methods are a middle ground between the simplicity of spectrophotometric methods and the variety of applications of biological sample radioassays. However, similarly to in the case of spectrophotometric methods, the use of non-natural substrates probably limits the activity of the enzymes. In fact, the spectrophotometric and fluorescent methods are predominantly used to control the loss of activity of the enzyme during purification steps. Therefore, despite the time and cost involved, radioactive assays appear to be the method of choice when it comes to actually measuring enzyme activity.
This entry is adapted from the peer-reviewed paper 10.3390/ijms231911093
References
- Long, J.Z.; Cravatt, B.F. The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chem. Rev. 2011, 111, 6022–6063.
- Zechner, R.; Kienesberger, P.C.; Haemmerle, G.; Zimmermann, R.; Lass, A. Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores. J. Lipid Res. 2009, 50, 3–21.
- Zimmermann, R.; Strauss, J.G.; Haemmerle, G.; Schoiswohl, G.; Birner-Gruenberger, R.; Riederer, M.; Lass, A.; Neuberger, G.; Eisenhaber, F.; Hermetter, A.; et al. Fat Mobilization in Adipose Tissue is Promoted by Adopose Triglyceride Lipase. Science 2004, 306, 1383–1386.
- Zimmermann, R.; Lass, A.; Haemmerle, G.; Zechner, R. Fate of fat: The role of adipose triglyceride lipase in lipolysis. Biochim. Biophys. Acta 2009, 1791, 494–500.
- Obrowsky, S.; Chandak, P.G.; Patankar, J.V.; Povoden, S.; Schlager, S.; Kershaw, E.E.; Bogner-Strauss, J.G.; Hoefler, G.; Levak-Frank, S.; Kratky, D. Adipose triglyceride lipase is a TG hydrolase of the small intestine and regulates intestinal PPARalpha signaling. J. Lipid Res. 2013, 54, 425–435.
- Liu, S.; Promes, J.A.; Harata, M.; Mishra, A.; Stephens, S.B.; Taylor, E.B.; Burand, A.J., Jr.; Sivitz, W.I.; Fink, B.D.; Ankrum, J.A.; et al. Adipose Triglyceride Lipase Is a Key Lipase for the Mobilization of Lipid Droplets in Human beta-Cells and Critical for the Maintenance of Syntaxin 1a Levels in beta-Cells. Diabetes 2020, 69, 1178–1192.
- Cerk, I.K.; Wechselberger, L.; Oberer, M. Adipose Triglyceride Lipase Regulation: An Overview. Curr. Protein Pept. Sci. 2018, 19, 221–233.
- Lass, A.; Zimmermann, R.; Haemmerle, G.; Riederer, M.; Schoiswohl, G.; Schweiger, M.; Kienesberger, P.; Strauss, J.G.; Gorkiewicz, G.; Zechner, R. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab. 2006, 3, 309–319.
- Kimmel, A.R.; Sztalryd, C. The Perilipins: Major Cytosolic Lipid Droplet-Associated Proteins and Their Roles in Cellular Lipid Storage, Mobilization, and Systemic Homeostasis. Annu. Rev. Nutr. 2016, 36, 471–509.
- Granneman, J.G.; Moore, H.P.; Mottillo, E.P.; Zhu, Z.; Zhou, L. Interactions of perilipin-5 (Plin5) with adipose triglyceride lipase. J. Biol. Chem. 2011, 286, 5126–5135.
- Sztalryd, C.; Brasaemle, D.L. The perilipin family of lipid droplet proteins: Gatekeepers of intracellular lipolysis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1221–1232.
- Yang, X.; Lu, X.; Lombes, M.; Rha, G.B.; Chi, Y.I.; Guerin, T.M.; Smart, E.J.; Liu, J. The G(0)/G(1) switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab. 2010, 11, 194–205.
- Nagy, H.M.; Paar, M.; Heier, C.; Moustafa, T.; Hofer, P.; Haemmerle, G.; Lass, A.; Zechner, R.; Oberer, M.; Zimmermann, R. Adipose triglyceride lipase activity is inhibited by long-chain acyl-coenzyme A. Biochim. Biophys. Acta 2014, 1841, 588–594.
- Mayer, N.; Schweiger, M.; Romauch, M.; Grabner, G.F.; Eichmann, T.O.; Fuchs, E.; Ivkovic, J.; Heier, C.; Mrak, I.; Lass, A.; et al. Development of small-molecule inhibitors targeting adipose triglyceride lipase. Nat. Chem. Biol. 2013, 9, 785–787.
- Iglesias, J.; Lamontagne, J.; Erb, H.; Gezzar, S.; Zhao, S.; Joly, E.; Truong, V.L.; Skorey, K.; Crane, S.; Madiraju, S.R.; et al. Simplified assays of lipolysis enzymes for drug discovery and specificity assessment of known inhibitors. J. Lipid Res. 2016, 57, 131–141.
- Grabner, G.F.; Guttenberger, N.; Mayer, N.; Migglautsch-Sulzer, A.K.; Lembacher-Fadum, C.; Fawzy, N.; Bulfon, D.; Hofer, P.; Zullig, T.; Hartig, L.; et al. Small-Molecule Inhibitors Targeting Lipolysis in Human Adipocytes. J. Am. Chem. Soc. 2022, 144, 6237–6250.
- Reisenberg, M.; Singh, P.K.; Williams, G.; Doherty, P. The diacylglycerol lipases: Structure, regulation and roles in and beyond endocannabinoid signalling. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 3264–3275.
- Kridel, S.J.; Axelrod, F.; Rozenkrantz, N.; Smith, J.W. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res.. 2004, 64, 2070–2075.
- McNeely, W.; Benfield, P. Orlistat. Drugs 1998, 56, 241–249, discussion 250.
- Sjöström, L.; Rissanen, A.; Andersen, T.; Boldrin, M.; Golay, A.; Koppeschaar, H.P.F.; Krempf, M. Randomised placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. Lancet 1998, 352, 167–172.
- Ballinger, A.; Peikin, S.R. Orlistat: Its current status as an anti-obesity drug. Eur. J. Pharmacol. 2001, 440, 109–117.
- Korbelius, M.; Vujic, N.; Sachdev, V.; Obrowsky, S.; Rainer, S.; Gottschalk, B.; Graier, W.F.; Kratky, D. ATGL/CGI-58-Dependent Hydrolysis of a Lipid Storage Pool in Murine Enterocytes. Cell Rep. 2019, 28, 1923–1934.e4.
- Cornaciu, I.; Boeszoermenyi, A.; Lindermuth, H.; Nagy, H.M.; Cerk, I.K.; Ebner, C.; Salzburger, B.; Gruber, A.; Schweiger, M.; Zechner, R.; et al. The minimal domain of adipose triglyceride lipase (ATGL) ranges until leucine 254 and can be activated and inhibited by CGI-58 and G0S2, respectively. PLoS ONE 2011, 6, e26349.
- Filleur, S.; Nelius, T.; de Riese, W.; Kennedy, R.C. Characterization of PEDF: A multi-functional serpin family protein. J. Cell. Biochem. 2009, 106, 769–775.
- Chen, Y.; Carlessi, R.; Walz, N.; Cruzat, V.F.; Keane, K.; John, A.N.; Jiang, F.X.; Carnagarin, R.; Dass, C.R.; Newsholme, P. Pigment epithelium-derived factor (PEDF) regulates metabolism and insulin secretion from a clonal rat pancreatic beta cell line BRIN-BD11 and mouse islets. Mol. Cell. Endocrinol. 2016, 426, 50–60.
- Kulminskaya, N.; Oberer, M. Protein-protein interactions regulate the activity of Adipose Triglyceride Lipase in intracellular lipolysis. Biochimie 2020, 169, 62–68.
- Kimmel, A.R.; Sztalryd, C. Perilipin 5, a lipid droplet protein adapted to mitochondrial energy utilization. Curr. Opin. Lipidol. 2014, 25, 110–117.
- Zagani, R.; El-Assaad, W.; Gamache, I.; Teodoro, J.G. Inhibition of adipose triglyceride lipase (ATGL) by the putative tumor suppressor G0S2 or a small molecule inhibitor attenuates the growth of cancer cells. Oncotarget 2015, 6, 28282–28295.
- Wan, Z.; Matravadia, S.; Holloway, G.P.; Wright, D.C. FAT/CD36 regulates PEPCK expression in adipose tissue. Am. J. Physiol. Cell Physiol. 2013, 304, C478–C484.
- de Oliveira, C.; Khatua, B.; Noel, P.; Kostenko, S.; Bag, A.; Balakrishnan, B.; Patel, K.S.; Guerra, A.A.; Martinez, M.N.; Trivedi, S.; et al. Pancreatic triglyceride lipase mediates lipotoxic systemic inflammation. J. Clin. Investig. 2020, 130, 1931–1947.
- Fako, V.E.; Zhang, J.T.; Liu, J.Y. Mechanism of Orlistat Hydrolysis by the Thioesterase of Human Fatty Acid Synthase. ACS Catal. 2014, 4, 3444–3453.
- Yeaman, S.J. Hormone-sensitive lipase--new roles for an old enzyme. Biochem. J. 2004, 379, 11–22.
- Mita, T.; Furuhashi, M.; Hiramitsu, S.; Ishii, J.; Hoshina, K.; Ishimura, S.; Fuseya, T.; Watanabe, Y.; Tanaka, M.; Ohno, K.; et al. FABP4 is secreted from adipocytes by adenyl cyclase-PKA- and guanylyl cyclase-PKG-dependent lipolytic mechanisms. Obesity 2015, 23, 359–367.
- Lafontan, M.; Langin, D. Lipolysis and lipid mobilization in human adipose tissue. Prog. Lipid Res. 2009, 48, 275–297.
- Holm, C. Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Biochem. Soc. Trans. 2003, 31, 1120–1124.
- Watt, M.J.; Holmes, A.G.; Pinnamaneni, S.K.; Garnham, A.P.; Steinberg, G.R.; Kemp, B.E.; Febbraio, M.A. Regulation of HSL serine phosphorylation in skeletal muscle and adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E500–E508.
- Viollet, B.; Andreelli, F.; Jorgensen, S.B.; Perrin, C.; Flamez, D.; Mu, J.; Wojtaszewski, J.F.; Schuit, F.C.; Birnbaum, M.; Richter, E.; et al. Physiological role of AMP-activated protein kinase (AMPK): Insights from knockout mouse models. Biochem. Soc. Trans. 2003, 31, 216–219.
- Ueno, M.; Suzuki, J.; Hirose, M.; Sato, S.; Imagawa, M.; Zenimaru, Y.; Takahashi, S.; Ikuyama, S.; Koizumi, T.; Konoshita, T.; et al. Cardiac overexpression of perilipin 2 induces dynamic steatosis: Prevention by hormone-sensitive lipase. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E699–E709.
- Claus, T.H.; Lowe, D.B.; Liang, Y.; Salhanick, A.I.; Lubeski, C.K.; Yang, L.; Lemoine, L.; Zhu, J.; Clairmont, K.B. Specific inhibition of hormone-sensitive lipase improves lipid profile while reducing plasma glucose. J. Pharmacol. Exp. Ther. 2005, 315, 1396–1402.
- Mottillo, E.P.; Shen, X.J.; Granneman, J.G. Role of hormone-sensitive lipase in beta-adrenergic remodeling of white adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1188–E1197.
- Gao, H.; Feng, X.J.; Li, Z.M.; Li, M.; Gao, S.; He, Y.H.; Wang, J.J.; Zeng, S.Y.; Liu, X.P.; Huang, X.Y.; et al. Downregulation of adipose triglyceride lipase promotes cardiomyocyte hypertrophy by triggering the accumulation of ceramides. Arch Biochem. Biophys. 2015, 565, 76–88.
- Ebdrup, ø.; Refsgaard, H.H.F.; Fledelius, C.; Jacobsen, P. Synthesis and Structure-Activity Relationship for a Novel Class of Potent and Selective Carbamate-Based Inhibitors of Hormone Selective Lipase with Acute In Vivo Antilipolytic Effects. J. Med. Chem. 2006, 2007, 5449–5456.
- Bustanji, Y.; Issa, A.; Mohammad, M.; Hudaib, M.; Tawah, K.; Alkhatib, H.; Almasri, I.; Al-Khalidi, B. Inhibition of hormone sensitive lipase and pancreatic lipase by Rosmarinus officinalis extract and selected phenolic constituents. J. Med. Plants Res. 2010, 4, 2235–2242.
- Vagelos, P.R.; Alberts, A.W.; Martin, D.B. Studies on the Mechanism of Activation of Acetyl Coenzyme A Carboxylase by Citrate. J. Biol. Chem. 1963, 238, 533–540.
- Beaty, N.B.; Lane, M.D. Kinetics of activation of acetyl-CoA carboxylase by citrate. Relationship to the rate of polymerization of the enzyme. J. Biol. Chem. 1983, 258, 13043–13050.
- Hardie, D.G.; Cohen, P. Purification and physicochemical properties of fatty acid synthetase and acetyl-CoA carboxylase from lactating rabbit mammary gland. Eur. J. Biochem. 1978, 92, 25–34.
- Boone, A.N.; Chan, A.; Kulpa, J.E.; Brownsey, R.W. Bimodal activation of acetyl-CoA carboxylase by glutamate. J. Biol. Chem. 2000, 275, 10819–10825.
- Kowluru, A.; Chen, H.-Q.; Modrick, L.M.; Stefanelli, C. Activation of Acetyl-CoA Carboxylase by a Glutamateand Magnesium-Sensitive Protein Phosphatase in the Islet β-Cell. Diabetes 2001, 50, 1580–1587.
- Brownsey, R.W.; Boone, A.N.; Elliott, J.E.; Kulpa, J.E.; Lee, W.M. Regulation of acetyl-CoA carboxylase. Biochem. Soc. Trans. 2006, 34, 223–227.
- Galic, S.; Loh, K.; Murray-Segal, L.; Steinberg, G.R.; Andrews, Z.B.; Kemp, B.E. AMPK signaling to acetyl-CoA carboxylase is required for fasting- and cold-induced appetite but not thermogenesis. Elife 2018, 7, e32656.
- Levert, K.L.; Waldrop, G.L.; Stephens, J.M. A biotin analog inhibits acetyl-CoA carboxylase activity and adipogenesis. J. Biol. Chem. 2002, 277, 16347–16350.
- Munday, M.R. Regulation of mammalian acetyl-CoA carboxylase. Biochem. Soc. Trans. 2002, 30, 1059–1064.
- Harwood, H.J., Jr.; Petras, S.F.; Shelly, L.D.; Zaccaro, L.M.; Perry, D.A.; Makowski, M.R.; Hargrove, D.M.; Martin, K.A.; Tracey, W.R.; Chapman, J.G.; et al. Isozyme-nonselective N-substituted bipiperidylcarboxamide acetyl-CoA carboxylase inhibitors reduce tissue malonyl-CoA concentrations, inhibit fatty acid synthesis, and increase fatty acid oxidation in cultured cells and in experimental animals. J. Biol. Chem. 2003, 278, 37099–37111.
- Chen, L.; Duan, Y.; Wei, H.; Ning, H.; Bi, C.; Zhao, Y.; Qin, Y.; Li, Y. Acetyl-CoA carboxylase (ACC) as a therapeutic target for metabolic syndrome and recent developments in ACC1/2 inhibitors. Expert Opin. Investig. Drugs 2019, 28, 917–930.
- Gerth, K.; Bedorf, N.; Irschik, H.; Hofle, G.; Reichenbach, H. The soraphens: A family of novel antifungal compounds from Sorangium cellulosum (Myxobacteria). I. Soraphen A1 alpha: Fermentation, isolation, biological properties. J. Antibiot. 1994, 47, 23–31.
- Schreurs, M.; van Dijk, T.H.; Gerding, A.; Havinga, R.; Reijngoud, D.J.; Kuipers, F. Soraphen, an inhibitor of the acetyl-CoA carboxylase system, improves peripheral insulin sensitivity in mice fed a high-fat diet. Diabetes Obes. Metab. 2009, 11, 987–991.
- Jump, D.B.; Torres-Gonzalez, M.; Olson, L.K. Soraphen A, an inhibitor of acetyl CoA carboxylase activity, interferes with fatty acid elongation. Biochem. Pharmacol. 2011, 81, 649–660.
- Harwood, H.J., Jr. Treating the metabolic syndrome: Acetyl-CoA carboxylase inhibition. Expert Opin. Ther. Targets 2005, 9, 267–281.
- Beckers, A.; Organe, S.; Timmermans, L.; Scheys, K.; Peeters, A.; Brusselmans, K.; Verhoeven, G.; Swinnen, J.V. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res. 2007, 67, 8180–8187.
- Harriman, G.; Greenwood, J.; Bhat, S.; Huang, X.; Wang, R.; Paul, D.; Tong, L.; Saha, A.K.; Westlin, W.F.; Kapeller, R.; et al. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc. Natl. Acad. Sci. USA 2016, 113, E1796–E1805.
- Li, E.Q.; Zhao, W.; Zhang, C.; Qin, L.Z.; Liu, S.J.; Feng, Z.Q.; Wen, X.; Chen, C.P. Synthesis and anti-cancer activity of ND-646 and its derivatives as acetyl-CoA carboxylase 1 inhibitors. Eur. J. Pharm. Sci. 2019, 137, 105010.
- Svensson, R.U.; Parker, S.J.; Eichner, L.J.; Kolar, M.J.; Wallace, M.; Brun, S.N.; Lombardo, P.S.; Van Nostrand, J.L.; Hutchins, A.; Vera, L.; et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat. Med. 2016, 22, 1108–1119.
- Bourbeau, M.P.; Bartberger, M.D. Recent advances in the development of acetyl-CoA carboxylase (ACC) inhibitors for the treatment of metabolic disease. J. Med. Chem. 2015, 58, 525–536.
- Esler, W.P.; Tesz, G.J.; Hellerstein, M.K.; Beysen, C.; Sivamani, R.; Turner, S.M.; Watkins, S.M.; Amor, P.A.; Carvajal-Gonzalez, S.; Geoly, F.J.; et al. Human sebum requires de novo lipogenesis, which is increased in acne vulgaris and suppressed by acetyl-CoA carboxylase inhibition. Sci. Transl. Med. 2019, 11, eaau8465.
- Huard, K.; Smith, A.C.; Cappon, G.; Dow, R.L.; Edmonds, D.J.; El-Kattan, A.; Esler, W.P.; Fernando, D.P.; Griffith, D.A.; Kalgutkar, A.S.; et al. Optimizing the Benefit/Risk of Acetyl-CoA Carboxylase Inhibitors through Liver Targeting. J. Med. Chem. 2020, 63, 10879–10896.
- Ryder, T.F.; Bergman, A.; King-Ahmad, A.; Amin, N.B.; Lall, M.S.; Ballard, T.E.; Kalgutkar, A.S. Pharmacokinetics, mass balance, metabolism, and excretion of the liver-targeted acetyl-CoA carboxylase inhibitor PF-05221304 (clesacostat) in humans. Xenobiotica 2022, 52, 240–253.
- Huang, H.; McIntosh, A.L.; Martin, G.G.; Petrescu, A.D.; Landrock, K.K.; Landrock, D.; Kier, A.B.; Schroeder, F. Inhibitors of Fatty Acid Synthesis Induce PPAR alpha -Regulated Fatty Acid beta -Oxidative Genes: Synergistic Roles of L-FABP and Glucose. PPAR Res. 2013, 2013, 865604.
- Wang, C.; Xu, C.; Sun, M.; Luo, D.; Liao, D.F.; Cao, D. Acetyl-CoA carboxylase-alpha inhibitor TOFA induces human cancer cell apoptosis. Biochem. Biophys. Res. Commun. 2009, 385, 302–306.
- Thupari, J.N.; Pinn, M.L.; Kuhajda, F.P. Fatty acid synthase inhibition in human breast cancer cells leads to malonyl-CoA-induced inhibition of fatty acid oxidation and cytotoxicity. Biochem. Biophys. Res. Commun. 2001, 285, 217–223.
- Kim, K.W.; Yamane, H.; Zondlo, J.; Busby, J.; Wang, M. Expression, purification, and characterization of human acetyl-CoA carboxylase 2. Protein Expr. Purif. 2007, 53, 16–23.
- Pizer, E.S.; Jackisch, C.; Wood, F.D.; Pasternack, G.R.; Davidson, N.E.; Kuhajda, F.P. Inhibition of fatty acid synthesis induces programmed cell death in human breast cancer cells. Cancer Res. 1996, 56, 2745–2747.
- Pizer, E.S.; Chrest, F.J.; DiGiuseppe, J.A.; Han, W.F. Pharmacological inhibitors of mammalian fatty acid synthase suppress DNA replication and induce apoptosis in tumor cell lines. Cancer Res. 1998, 58, 4611–4615.
- Vance, D.; Omura, S.; Nomura, S.; Mitsuhashi, O.; Bloch, K.; Goldberg, I. Inhibition of Fatty-Acid Synthetases by Antibiotic Cerulenin. Biochem. Biophys. Res. Commun. 1972, 48, 649–656.
- Vazquez-Martin, A.; Ropero, S.; Brunet, J.; Colomer, R.; Menendez, J.A. Inhibition of Fatty Acid Synthase (FASN) synergistically enhances the efficacy of 5-fluorouracil in breast carcinoma cells. Oncol. Rep. 2007, 18, 973–980.
- Johansson, P.; Wiltschi, B.; Kumari, P.; Kessler, B.; Vonrhein, C.; Vonck, J.; Oesterhelt, D.; Grininger, M. Inhibition of the fungal fatty acid synthase type I multienzyme complex. Proc. Natl. Acad. Sci. USA 2008, 105, 12803–12808.
- Rudolph, M.C.; Karl Maluf, N.; Wellberg, E.A.; Johnson, C.A.; Murphy, R.C.; Anderson, S.M. Mammalian fatty acid synthase activity from crude tissue lysates tracing (1)(3)C-labeled substrates using gas chromatography-mass spectrometry. Anal. Biochem. 2012, 428, 158–166.
- Kuhajda, F.P.; Pizer, E.S.; Li, J.N.; Mani, N.S.; Frehywot, G.L.; Townsend, C.A. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc. Natl. Acad. Sci. USA 2000, 97, 3450–3454.
- Rendina, A.R.; Cheng, D. Characterization of the inactivation of rat fatty acid synthase by C75: Inhibition of partial reactions and protection by substrates. Biochem. J. 2005, 388, 895–903.
- Boelcke, W.P.; Teixeira, I.F.; Aquino, I.G.; Mazzaro, A.R.; Cuadra-Zelaya, F.J.M.; de Souza, A.P.; Salo, T.; Della Coletta, R.; Graner, E.; Bastos, D.C. Pharmacological fatty acid synthase inhibitors differently affect the malignant phenotype of oral cancer cells. Arch. Oral Biol. 2022, 135, 105343.
- Liu, H.; Liu, J.-Y.; Wu, X.; Zhang, J.-T. Biochemistry, molecular biology, and pharmacology of fatty acid synthase, an emerging therapeutic target and diagnosis/prognosis marke. Int. J. Biochem. Mol. Biol. 2010, 1, 69–89.
- Pemble, C.W.t.; Johnson, L.C.; Kridel, S.J.; Lowther, W.T. Crystal structure of the thioesterase domain of human fatty acid synthase inhibited by Orlistat. Nat. Struct. Mol. Biol. 2007, 14, 704–709.
- Zhang, J.S.; Lei, J.P.; Wei, G.Q.; Chen, H.; Ma, C.Y.; Jiang, H.Z. Natural fatty acid synthase inhibitors as potent therapeutic agents for cancers: A review. Pharm. Biol. 2016, 54, 1919–1925.
- Liu, B.; Wang, Y.; Fillgrove, K.L.; Anderson, V.E. Triclosan inhibits enoyl-reductase of type I fatty acid synthase in vitro and is cytotoxic to MCF-7 and SKBr-3 breast cancer cells. Cancer Chemother. Pharmacol. 2002, 49, 187–193.
- Stewart, M.J.; Parikh, S.; Xiao, G.; Tonge, P.J.; Kisker, C. Structural basis and mechanism of enoyl reductase inhibition by triclosan. J. Mol. Biol. 1999, 290, 859–865.
- Sun, D.; Zhao, T.; Long, K.; Wu, M.; Zhang, Z. Triclosan down-regulates fatty acid synthase through microRNAs in HepG2 cells. Eur. J. Pharmacol. 2021, 907, 174261.
- Sadowski, M.C.; Pouwer, R.H.; Gunter, J.H.; Lubik, A.A.; Quinn, R.J.; Nelson, C.C. The fatty acid synthase inhibitor triclosan: Repurposing an antimicrobial agent for targeting prostate cancer. Oncotarget 2014, 5, 9362–9381.
- Recazens, E.; Mouisel, E.; Langin, D. Hormone-sensitive lipase: Sixty years later. Prog. Lipid Res. 2021, 82, 101084.
- Bezaire, V.; Mairal, A.; Anesia, R.; Lefort, C.; Langin, D. Chronic TNFalpha and cAMP pre-treatment of human adipocytes alter HSL, ATGL and perilipin to regulate basal and stimulated lipolysis. FEBS Lett. 2009, 583, 3045–3049.
- Langin, D.; Dicker, A.; Tavernier, G.; Hoffstedt, J.; Mairal, A.; Ryden, M.; Arner, E.; Sicard, A.; Jenkins, C.M.; Viguerie, N.; et al. Adipocyte lipases and defect of lipolysis in human obesity. Diabetes 2005, 54, 3190–3197.
- Agustsson, T.; Ryden, M.; Hoffstedt, J.; van Harmelen, V.; Dicker, A.; Laurencikiene, J.; Isaksson, B.; Permert, J.; Arner, P. Mechanism of increased lipolysis in cancer cachexia. Cancer Res. 2007, 67, 5531–5537.
- Kim, K.; Kang, J.K.; Jung, Y.H.; Lee, S.B.; Rametta, R.; Dongiovanni, P.; Valenti, L.; Pajvani, U.B. Adipocyte PHLPP2 inhibition prevents obesity-induced fatty liver. Nat. Commun. 2021, 12, 1822.
- Schaffer, J.E. Lipotoxicity: When tissues overeat. Curr. Opin. Lipidol. 2003, 14, 281–287.
- Heier, C.; Knittelfelder, O.; Hofbauer, H.F.; Mende, W.; Pornbacher, I.; Schiller, L.; Schoiswohl, G.; Xie, H.; Gronke, S.; Shevchenko, A.; et al. Hormone-sensitive lipase couples intergenerational sterol metabolism to reproductive success. Elife 2021, 10, e63252.
- Shiau, M.Y.; Chuang, P.H.; Yang, C.P.; Hsiao, C.W.; Chang, S.W.; Chang, K.Y.; Liu, T.M.; Chen, H.W.; Chuang, C.C.; Yuan, S.Y.; et al. Mechanism of Interleukin-4 Reducing Lipid Deposit by Regulating Hormone-Sensitive Lipase. Sci. Rep. 2019, 9, 11974.
- Arredondo-Amador, M.; Zambrano, C.; Kulyte, A.; Lujan, J.; Hu, K.; Sanchez de Medina, F.; Scheer, F.; Arner, P.; Ryden, M.; Martinez-Augustin, O.; et al. Circadian Rhythms in Hormone-sensitive Lipase in Human Adipose Tissue: Relationship to Meal Timing and Fasting Duration. J. Clin. Endocrinol. Metab. 2020, 105, e4407–e4416.
- Zhuan, Q.; Ma, H.; Chen, J.; Luo, Y.; Luo, Y.; Gao, L.; Hou, Y.; Zhu, S.; Fu, X. Cytoplasm lipids can be modulated through hormone-sensitive lipase and are related to mitochondrial function in porcine IVM oocytes. Reprod. Fertil. Dev. 2020, 32, 667–675.
- Schweiger, M.; Eichmann, T.O.; Taschler, U.; Zimmermann, R.; Zechner, R.; Lass, A. Measurement of lipolysis. Methods Enzymol. 2014, 538, 171–193.
- Holm, C.; Contreras, J.A.; Verger, R.; Schotz, M.C. Large-scale purification and kinetic properties of recombinant hormone-sensitive lipase from baculovirus-insect cell systems. Methods Enzymol. 1997, 284, 272–284.
- Steinberg, G.R.; Kemp, B.E.; Watt, M.J. Adipocyte triglyceride lipase expression in human obesity. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E958–E964.
- Schweiger, M.; Schreiber, R.; Haemmerle, G.; Lass, A.; Fledelius, C.; Jacobsen, P.; Tornqvist, H.; Zechner, R.; Zimmermann, R. Adipose triglyceride lipase and hormone-sensitive lipase are the major enzymes in adipose tissue triacylglycerol catabolism. J. Biol. Chem. 2006, 281, 40236–40241.
- Haemmerle, G.; Zimmermann, R.; Hayn, M.; Theussl, C.; Waeg, G.; Wagner, E.; Sattler, W.; Magin, T.M.; Wagner, E.F.; Zechner, R. Hormone-sensitive lipase deficiency in mice causes diglyceride accumulation in adipose tissue, muscle, and testis. J. Biol. Chem. 2002, 277, 4806–4815.
- Fredrikson, G.; Strålfors, P.; Nilsson, N.Ö.; Belfrage, P. Hormone-Sensitive Lipase from Adipose Tissue of Rat. Methods Enzymol. 1981, 71, 636–646.
- Watt, M.J.; Carey, A.L.; Wolsk-Petersen, E.; Kraemer, F.B.; Pedersen, B.K.; Febbraio, M.A. Hormone-sensitive lipase is reduced in the adipose tissue of patients with type 2 diabetes mellitus: Influence of IL-6 infusion. Diabetologia 2005, 48, 105–112.
- Watt, M.J.; Heigenhauser, G.J.; Spriet, L.L. Effects of dynamic exercise intensity on the activation of hormone-sensitive lipase in human skeletal muscle. J. Physiol. 2003, 547, 301–308.
- Holm, C.; Langin, D.; Manganiello, V.; Belfrage, P.; Degerman, E. Regulation of hormone-sensitive lipase activity in adipose tissue. Lipases 1997, 286, 45–67.
- Richelsen, B.; Pedersen, S.B.; Kristensen, K.; Borglum, J.D.; Norrelund, H.; Christiansen, J.S.; Jorgensen, J.O. Regulation of lipoprotein lipase and hormone-sensitive lipase activity and gene expression in adipose and muscle tissue by growth hormone treatment during weight loss in obese patients. Metabolism 2000, 49, 906–911.
- Schweiger, M.; Schoiswohl, G.; Lass, A.; Radner, F.P.; Haemmerle, G.; Malli, R.; Graier, W.; Cornaciu, I.; Oberer, M.; Salvayre, R.; et al. The C-terminal region of human adipose triglyceride lipase affects enzyme activity and lipid droplet binding. J. Biol. Chem. 2008, 283, 17211–17220.
- Takagi, A.; Ikeda, Y.; Kobayashi, K.; Kobayashi, K.; Ikeda, Y.; Kozawa, J.; Miyauchi, H.; Li, M.; Hashimoto, C.; Hara, Y.; et al. Newly developed selective immunoinactivation assay revealed reduction in adipose triglyceride lipase activity in peripheral leucocytes from patients with idiopathic triglyceride deposit cardiomyovasculopathy. Biochem. Biophys. Res. Commun. 2018, 495, 646–651.
- Chandak, P.G.; Radovic, B.; Aflaki, E.; Kolb, D.; Buchebner, M.; Frohlich, E.; Magnes, C.; Sinner, F.; Haemmerle, G.; Zechner, R.; et al. Efficient phagocytosis requires triacylglycerol hydrolysis by adipose triglyceride lipase. J. Biol. Chem. 2010, 285, 20192–20201.
- Reid, B.N.; Ables, G.P.; Otlivanchik, O.A.; Schoiswohl, G.; Zechner, R.; Blaner, W.S.; Goldberg, I.J.; Schwabe, R.F.; Chua, S.C., Jr.; Huang, L.S. Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis. J. Biol. Chem. 2008, 283, 13087–13099.
- Mersmann, H.J. Lipoprotein and hormone-sensitive lipases in porcine adipose tissue. J. Anim. Sci. 1998, 76, 1396–1404.
- Holm, C.; Olivecrona, G.; Ottosson, M. Assays of lipolytic enzymes. Methods Mol. Biol. 2001, 155, 97–119.
- Sekiya, M.; Osuga, J.; Yahagi, N.; Okazaki, H.; Tamura, Y.; Igarashi, M.; Takase, S.; Harada, K.; Okazaki, S.; Iizuka, Y.; et al. Hormone-sensitive lipase is involved in hepatic cholesteryl ester hydrolysis. J. Lipid Res. 2008, 49, 1829–1838.
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155.
- Ding, Y.; Zhang, S.; Yang, L.; Na, H.; Zhang, P.; Zhang, H.; Wang, Y.; Chen, Y.; Yu, J.; Huo, C.; et al. Isolating lipid droplets from multiple species. Nat. Protoc. 2013, 8, 43–51.
- Brasaemle, D.L.; Wolins, N.E. Isolation of Lipid Droplets from Cells by Density Gradient Centrifugation. Curr. Protoc. Cell Biol. 2016, 72, 3.15.1–3.15.13.
- Borg, M.L.; Andrews, Z.B.; Duh, E.J.; Zechner, R.; Meikle, P.J.; Watt, M.J. Pigment epithelium-derived factor regulates lipid metabolism via adipose triglyceride lipase. Diabetes 2011, 60, 1458–1466.
- Bradley, D.C.; Kaslow, H.R. Radiometric assays for glycerol, glucose, and glycogen. Anal. Biochem. 1989, 180, 11–16.
- Slavin, B.G.; Ong, J.M.; Kern, P.A. Hormonal regulation of hormone-sensitive lipase activity and mRNA levels in isolated rat adipocytes. J. Lipid Res. 1994, 35, 1535–1541.
- Amols, H.I.; Zaider, M.; Weinberger, J.; Ennis, R.; Schiff, P.B.; Reinstein, L.E. Dosimetric considerations for catheter-based beta and gamma emitters in the therapy of neointimal hyperplasia in human coronary arteries. Int. J. Radiat. Oncol. Biol. Phys. 1996, 36, 913–921.
- Morak, M.; Schmidinger, H.; Riesenhuber, G.; Rechberger, G.N.; Kollroser, M.; Haemmerle, G.; Zechner, R.; Kronenberg, F.; Hermetter, A. Adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL) deficiencies affect expression of lipolytic activities in mouse adipose tissues. Mol. Cell. Proteom. 2012, 11, 1777–1789.
- Witters, L.A.; Watts, T.D.; Daniels, D.L.; Evans, J.L. Insulin stimulates the dephosphorylation and activation of acetyl-CoA carboxylase. Proc. Natl. Acad. Sci. USA 1988, 85, 5473–5477.
- Gregolin, C.; Ryder, E.; Kleinschmidt, A.K.; Warner, R.C.; Lane, M.D. Molecular characteristics of liver acetyl CoA carboxylase. Proc. Natl. Acad. Sci. USA 1966, 56, 148–155.
- Dean, D.; Daugaard, J.R.; Young, M.E.; Saha, A.; Vavvas, D.; Asp, S.; Kiens, B.; Kim, K.H.; Witters, L.; Richter, E.A.; et al. Exercise diminishes the activity of acetyl-CoA carboxylase in human muscle. Diabetes 2000, 49, 1295–1300.
- Moibi, J.A.; Ekpe, E.D.; Christopherson, R.J. Acetyl-CoA carboxylase and fatty acid synthase activity and immunodetectable protein in adipose tissues of ruminants: Effect of temperature and feeding level. J. Anim. Sci. 2000, 78, 2383–2392.
- Stansbie, D.; Brownsey, R.W.; Crettaz, M.; Denton, R.M. Acute effects in vivo of anti-insulin serum on rates of fatty acid synthesis and activities of acetyl-coenzyme A carboxylase and pyruvate dehydrogenase in liver and epididymal adipose tissue of fed rats. Biochem. J. 1976, 160, 413–416.
- Halestrap, A.P.; Denton, R.M. Hormonal regulation of adipose-tissue acetyl-Coenzyme A carboxylase by changes in the polymeric state of the enzyme. The role of long-chain fatty acyl-Coenzyme A thioesters and citrate. Biochem. J. 1974, 142, 365–377.
- Bijleveld, C.; Geelen, M.J. Measurement of acetyl-CoA carboxylase activity in isolated hepatocytes. Biochim. Biophys. Acta 1987, 918, 274–283.
- Witters, L.A.; Moriarity, D.; Martin, D.B. Regulation of hepatic acetyl coenzyme A carboxylase by insulin and glucagon. J. Biol. Chem. 1979, 254, 6644–6649.
- Thampy, K.G.; Wakil, S.J. Activation of acetyl-CoA carboxylase. Purification and properties of a Mn2+-dependent phosphatase. J. Biol. Chem. 1985, 260, 6318–6323.
- Wong, K.; Meyers, R.; Cantley, L.C. Subcellular locations of phosphatidylinositol 4-kinase isoforms. J. Biol. Chem. 1997, 272, 13236–13241.
- Zimmermann, R.; Haemmerle, G.; Wagner, E.M.; Strauss, J.G.; Kratky, D.; Zechner, R. Decreased fatty acid esterification compensates for the reduced lipolytic activity in hormone-sensitive lipase-deficient white adipose tissue. J. Lipid Res. 2003, 44, 2089–2099.
- Chung, C.C.; Ohwaki, K.; Schneeweis, J.E.; Stec, E.; Varnerin, J.P.; Goudreau, P.N.; Chang, A.; Cassaday, J.; Yang, L.; Yamakawa, T.; et al. A fluorescence-based thiol quantification assay for ultra-high-throughput screening for inhibitors of coenzyme A production. Assay Drug Dev. Technol. 2008, 6, 361–374.
- Cerne, D.; Zitnik, I.P.; Sok, M. Increased fatty acid synthase activity in non-small cell lung cancer tissue is a weaker predictor of shorter patient survival than increased lipoprotein lipase activity. Arch. Med. Res. 2010, 41, 405–409.
- Topolska, M.; Martinez-Montanes, F.; Ejsing, C.S. A Simple and Direct Assay for Monitoring Fatty Acid Synthase Activity and Product-Specificity by High-Resolution Mass Spectrometry. Biomolecules 2020, 10, 118.
- Jayakumar, A.; Tai, M.H.; Huang, W.Y.; al-Feel, W.; Hsu, M.; Abu-Elheiga, L.; Chirala, S.S.; Wakil, S.J. Human fatty acid synthase: Properties and molecular cloning. Proc. Natl. Acad. Sci. USA 1995, 92, 8695–8699.
- Dils, R.; Carey, E.M. Fatty acid synthase from rabbit mammary gland. Methods Enzymol. 1975, 35, 74–83.
- Nepokroeff, C.M.; Lakshmanan, M.R.; Porter, J.W. Fatty-acid synthase from rat liver. Methods Enzymol. 1975, 35, 37–44.
- Alwarawrah, Y.; Hughes, P.; Loiselle, D.; Carlson, D.A.; Darr, D.B.; Jordan, J.L.; Xiong, J.; Hunter, L.M.; Dubois, L.G.; Thompson, J.W.; et al. Fasnall, a Selective FASN Inhibitor, Shows Potent Anti-tumor Activity in the MMTV-Neu Model of HER2(+) Breast Cancer. Cell Chem. Biol. 2016, 23, 678–688.
- Najjar, S.M.; Yang, Y.; Fernstrom, M.A.; Lee, S.J.; Deangelis, A.M.; Rjaily, G.A.; Al-Share, Q.Y.; Dai, T.; Miller, T.A.; Ratnam, S.; et al. Insulin acutely decreases hepatic fatty acid synthase activity. Cell Metab. 2005, 2, 43–53.
- Roncari, D.A. Fatty acid synthase from human liver. Methods Enzymol. 1981, 71, 73–79.
- Brusselmans, K.; Vrolix, R.; Verhoeven, G.; Swinnen, J.V. Induction of cancer cell apoptosis by flavonoids is associated with their ability to inhibit fatty acid synthase activity. J. Biol. Chem. 2005, 280, 5636–5645.
- Hsu, R.Y.; Butterworth, P.H.W.; Porter, J.W. Pigeon Liver Fatty Acid Synthase. Methods Enzymol. 1969, 14, 33–39.
- Moustaid, N.; Hainque, B.; Quignard-Boulange, A. Dexamethasone regulation of terminal differentiation in 3T3-F442A preadipocyte cell line. Cytotechnology 1988, 1, 285–293.
- Puig, T.; Turrado, C.; Benhamu, B.; Aguilar, H.; Relat, J.; Ortega-Gutierrez, S.; Casals, G.; Marrero, P.F.; Urruticoechea, A.; Haro, D.; et al. Novel Inhibitors of Fatty Acid Synthase with Anticancer Activity. Clin. Cancer Res. 2009, 15, 7608–7615.
- Lin, H.P.; Cheng, Z.L.; He, R.Y.; Song, L.; Tian, M.X.; Zhou, L.S.; Groh, B.S.; Liu, W.R.; Ji, M.B.; Ding, C.; et al. Destabilization of Fatty Acid Synthase by Acetylation Inhibits De Novo Lipogenesis and Tumor Cell Growth. Cancer Res. 2016, 76, 6924–6936.
- Moustaid, N.; Jones, B.H.; Taylor, J.W. Insulin increases lipogenic enzyme activity in human adipocytes in primary culture. J. Nutr. 1996, 126, 865–870.
- Wang, Y.; Jones Voy, B.; Urs, S.; Kim, S.; Soltani-Bejnood, M.; Quigley, N.; Heo, Y.R.; Standridge, M.; Andersen, B.; Dhar, M.; et al. The human fatty acid synthase gene and de novo lipogenesis are coordinately regulated in human adipose tissue. J. Nutr. 2004, 134, 1032–1038.
- Xue, B.; Zemel, M.B. Relationship between Human Adipose Tissue Agouti and Fatty Acid Synthase (FAS). J. Nutr. 2000, 130, 2478–2481.
This entry is offline, you can click here to edit this entry!