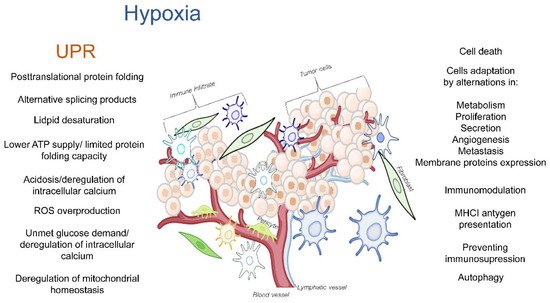
The tumor microenvironment (TME) is a dynamic network that is created by blood vessels, lymphatic vessels, fibroblasts, immune cells as well as components such as the extracellular matrix (ECM) that establishes a “friendly ecosystem” for cancer cells. Hypoxia induces a cellular adaptive response that elevates the expression of the transcription factors called hypoxia-inducible factors (HIFs) that activate the global gene expression changes in both non-malignant and cancer cells. While most of the studies in this area have focused on the canonical responses to hypoxia, a better understanding is needed for the complex molecular changes that are found in the hypoxic TME. These changes include the deregulation of endoplasmic reticulum (ER) homeostasis, and the subsequent perturbations in protein folding and secretion. The potential for erratic protein folding can also lead to another specialized stress response signaling pathway called the unfolded protein response (UPR). The UPR promotes survival during hypoxia by restoring the endoplasmic and mitochondrial homeostasis, but at times, it can also inhibit the cancer cell’s survival. The maturation of transmembrane and secretory proteins that include proangiogenic receptors and ligands as well as ECM remodeling enzymes takes place in the ER.
1. Hypoxia as an Activator of the UPR in the Tumor Microenvironment
Both hypoxia and the persistent deregulation of ER homeostasis during the activation of the UPR have been reported to be important features of the TME that affect both cancerous as well as non-malignant cells [
31,
46,
59,
60,
63,
64,
65,
66,
67,
68,
69,
70,
71,
72,
73,
74]. The constitutive activation of these pathways supports cancer cell survival through proliferation and by altering the innate and adaptive immune cells to promote the tumor’s progression and metastasis. Although the unmet oxygen demand does not dramatically restrict the disulfide bond formation during the protein synthesis, the posttranslational folding of the proteins is oxygen-dependent. Hypoxia limits the activity of the oxygen-dependent ER-localized oxidoreductase (ERO1α), and this leads to the deregulation of the posttranslational protein modifications and thereby, promotes ER stress [
75,
76]. Furthermore, exposure to hypoxia often results in the alternative splicing of several common proteins that can lead to the activation of UPR signaling [
77]. Notably, the lipid desaturation processes that are necessary for maintaining ER membrane homeostasis are oxygen-dependent as well [
78].
The cellular oxygen levels also influence the protein stability of the HIF transcription factors. Intracellular oxygen level-sensing mechanisms rely on the activity of the proline-hydroxylases (PHDs) and the asparaginyl-hydroxylase activity of factor-inhibiting HIF (FIH). During normoxic conditions, these hydroxylases post-translationally mark the HIF-α subunits for proteasomal degradation, and in doing so, they prevent the HIF transcriptional activity [
79,
80,
81,
82,
83,
84]. Interestingly, the HIF-β subunits are stable under these conditions [
79,
80,
81,
82,
83], thus indicating that the HIF regulation of the activity only occurs through the degradation of the alpha subunits. Hypoxia leads to the impairment of the PHDs and FIH activity and thus, an accumulation of the functional α-β-subunit HIFs complexes [
79,
80,
81,
82,
83] that are responsible for the extensive transcriptional reprograming of cellular functions that allow the cells to survive and adapt to this stress response. HIFs, through a direct interaction with the hypoxia response elements (HREs) consensus sequences in their target genes, modulate their levels in order to switch their metabolism to that of the less energy efficient non-oxidative mitochondrial activity [
9,
85,
86,
87,
88,
89].
One of the important functions of HIFs is to prevent the conversion of pyruvate to acetyl-Co-A and to increase the expression of glucose transporters and glycolytic enzymes to emphasize the glycolytic pathway [
90,
91,
92,
93]. The HIFs also down-regulate cytochrome c oxidase (COX) subunit composition expression [
94,
95], and this is accompanied by a HIF-elevated expression of carbonic anhydrase 9 (
CA-
IX) and monocarboxylate transporter 4 (
MCT4) that prevent the acidosis as a consequence of glycolysis-related proton release [
96,
97]. These glycolytic changes result in a lower ATP production, and the cellular energy requirements are limited by a HIF-mediated selective translational blockage [
98,
99,
100]. This also promotes the induction of autophagy and mitophagy [
100,
101,
102,
103] by an-mTOR independent pathway [
31,
104].
During this glycolytic switch, the cellular ATP-dependent processes such as protein synthesis, disulfide-bonds formation, peptide folding, and the maintenance of the redox potential and ion homeostasis are limited [
49,
105]. Furthermore, the hypoxia-related metabolic switch increases the production of lactic acid, thereby resulting in acidosis, the deregulation of intracellular calcium levels, and the overproduction of reactive oxygen species (ROS) [
106,
107,
108]. Limited the glucose and glutamine demands would reduce the synthesis of uridine diphosphate-
N-acetylglucosamine (UDP-GlcNAc) and thereby, limit the N-linked glycosylation in ER [
109] as well as deregulate the ER calcium influx [
110]. Furthermore, the changes in mitochondrial activity would result in the intracellular accumulation of ROS [
111] since high amounts of ROS are generated as a byproduct of fatty acid β-oxidation [
112,
113,
114]. Some of the TME deregulated cytokines and growth factors have also been reported to activate the NADPH oxidases and contribute to the ROS accumulation [
45]. Finally, a prolonged hypoxia disturbs the protein import processes in the mitochondria as well as mitochondrial protein folding, and this activates the mitochondrial unfolded protein response (UPRmt) [
115,
116,
117,
118]. Hence, hypoxia, along with all these other factors, can deregulate the cellular proteostasis and consequently activate the UPR. This results in a complex molecular network of interactions that affect the TME, and consequently, the tumor’s progression (
Figure 1).
Figure 1. The hypoxia-related deregulation of ER homeostasis in TME cells that can result in activation of the UPR and UPRmt and subsequently modulate TME.
Although HIF complexes containing HIF-1α subunits are considered to be the principal mediators of the cellular responses to hypoxia, in specific tissues, their functions can be supported and extended by the complexes that are formed by other α isoforms that include HIF-2α and HIF-3α [
9,
14,
119,
120,
121,
122,
123,
124]. The HIF-dependent transcriptional reprograming is not limited to a metabolic switch and facilitating cellular survival, but also to restoring oxygen homeostasis through promoting angiogenesis [
9,
13,
125].
2. The UPR and UPRmt
The occurrence of an ER stress increases the demand for chaperones in the lumen of this organelle, and this leads to UPR initiation which begins with the glucose-regulated protein 78 (GRP78 also known as BiP (binding immunoglobin protein)) release from the luminal domains of three ER transmembrane sensors: protein kinase RNA-like endoplasmic reticulum kinase (PERK), inositol-requiring enzyme 1α (IRE1α), and activating transcription factor 6 (ATF6) [
37,
42]. BiP dissociation activates PERK and IRE1α via multimerization and trans-autophosphorylation, and this allows the ATF6 proteolytic maturation into an active ATF6f (p50) transcription factor to occur [
138,
139,
140]. Upon their activation, all of these proteins initiate signaling pathways that function to help the cells to adapt to this insult, to repair the damage, and to restore ER homeostasis [
141]. The ATF6f promotes the synthesis of the protein chaperones (including BiP) and the ER membrane lipids, the ER-associated degradation (EDEM) of the misfolded proteins, and it enhances N-glycosylation [
142,
143]. IRE1α is also responsible for IRE1-dependent decay (RIDD) that degrades selected mRNAs in order to reduce the ER load [
144,
145,
146,
147] as well as IRE1 α splices the mRNA transcript of X-box binding-protein (XBP) transcription factor into its transcriptionally active isoform (
XBP1s) [
148]. XBP1s promote the ER membrane’s biosynthesis and support its folding capacity [
9,
36,
148,
149]. The main consequence of PERK activation is the phosphorylation of the alpha subunit of the eukaryotic initiation factor eIF2. This promotes a general suppression of protein synthesis [
42,
150,
151], and it allows for the increased expression of specific proteins including (1) growth arrest and DNA damage inducible protein (GADD34), (2) proapoptotic CCAAT/enhancer binding homologous protein (CHOP), and (3) activating transcription factor 4 (ATF4). ATF4 transcriptionally supports the adaptation to an ER stress and protein folding [
152], whereas GADD34 enables the dephosphorylation of eIF2, and thus removes the translational blockage when the ER stress is mitigated [
153].
In non-malignant cells, if the UPR stress response is too persistent or too intense, the cell death pathways are initiated. Although the accumulation of ATF4 and the PERK-dependent proapoptotic factor CHOP are well recognized as cell death signals, IRE1 can stimulate the Janus N-terminal kinase (JNK) to increase the expression of the death receptor 5 (
DR5), and ATF6f can also contribute to the CHOP accumulation [
9,
144,
154,
155]. The UPR is also accompanied by complex changes in many other apoptotic proteins such as the p53 upregulated modulator of apoptosis (PUMA), phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1, also known as NOXA), and growth arrest and DNA damage -inducible alpha GADD45A [
141,
146,
156,
157,
158,
159], which together with the main signals, influence the cell’s fate.
In the TME cancer cells, the UPR-specific signals are often clouded via the oncogenic transformations [
45] given that activation of the oncogenes results in an increased protein and membrane synthesis [
176]. Furthermore, the cancer cells adapt to avoid the UPR cell death signals as illustrated by the MYC Proto-Oncogene example. Increased levels of this oncogene in normal cells results in apoptosis, whereas in cancers cells, both the XBP1s and IRE1 allow for the cells to avoid an MYC-related cell death [
177,
178,
179]. The IRE1 pathway also modulates some of the effects of the mutant RAS, however, the significance of this for cancer cell survival remains unclear [
180].
Finally, the hypoxia-related metabolic switch and the resulting energetic deficiency may disrupt the mitochondrial protein homeostasis and lead to the activation of the mitochondrial UPR (UPRmt). This could occur via the limiting of the influx of the nuclear-encoded proteins and interfere with mitochondrial refolding after the protein import and activation of the UPRmt [
115,
116,
117,
118,
181]. The UPRmt is a proadaptive mechanism that changes the expression of both the mitochondrial and nuclear encoded genes (including
ATF5 and
ATF4) to restore homeostasis or if this fails, it leads to apoptosis [
115,
116,
117,
118]. Notably, the UPRmt-related activation of the PERK pathway has also been reported [
115,
116,
117,
118,
182].
3. The Crosstalk between Hypoxia and UPR in the TME
Although the pronounced activation of UPR signaling in cancer has been reported mainly for extreme hypoxic conditions [
65,
146], numerous reports have indicated that particular aspects of this are triggered even in less oxygen-limiting conditions, including increased BIP expression [
33,
123,
183,
184,
185,
186,
187] and PERK-related activity [
73,
184,
188,
189,
190,
191,
192]. The hypoxia-elevated BiP levels result from the activities of the extracellular signal-regulated kinase (ERK) and protein kinase C (PKC) [
184]. Furthermore, the increase in BIP expression can result from the hypoxic induction of its cofactor and transcriptional inducer called the cell migration-inducing and hyaluronan-binding ER protein (CEMIP) [
193]. In the ER, CEMIP/GRP78 support the adaptive responses by raising the intracellular calcium levels and subsequently increasing the PKCα activity [
193].
Although the PERK-dependent translational attenuation occurs immediately during acute hypoxia, this blockage is removed after a prolonged hypoxic exposure or with increased oxygen levels [
188,
194,
195]. The PERK-related eIF2 phosphorylation is also present in the transient (cyclic hypoxia) models [
196,
197,
198,
199,
200], and thus, the activation of this branch of UPR is very plausible in solid tumors that are characterized by fluctuating oxygen concentrations during tumor expansion. PERK activation inhibits the HIF-1α translation in cancer cells and thus, limits the HIF-1 transcriptional activity [
201].
The transcriptional activity of PERK-preferentially translated ATF4 is limited by PHD1, whereas ATF4 was shown to destabilize PHD3 and thus, support the HIF-1α accumulation [
202]. The question of whether this mechanism serves as a buffer of the HIF-1-related cellular adaptation or contributes to cell death will require further research.
Although PERK activation during hypoxia can lead to increased levels of CHOP expression and cell death in normal cells [
179,
207,
208,
209], in tumors, the CHOP expression is not elevated as dramatically as it is in the pharmacologically induced ER stress [
188]. Furthermore, CHOP can also serve in a proadaptive role by limiting the activity of the endothelial nitric synthase (
NOS3, eNOS) [
210] and preventing the ROS accumulation through the hypoxic uncoupling of this enzyme [
44,
211,
212].
Despite the fact that the PERK pathway has been considered as the main response pathway of the UPR in hypoxic tumors, the activation of the two other branches occurs as well. The elevated expression of ATF6-dependent prosurvival genes in response to hypoxia has been reported in gastric tumors [
213] and mutant p53 cancer cells [
214]. Furthermore, the elevated levels of these transcription factors are a hallmark of a poor prognosis for pancreatic cancer patients [
16]. The hypoxic activation of ATF6 signaling in the TME, however, has not been convincingly presented so far [
188].
Although elevated levels of XBP1s have been reported in many types of cancers and this has been correlated with poor prognosis, the IRE1 activity and the related accumulation of XBP1s has been reported mainly in cells that have been exposed acute and moderate hypoxia [
146,
188,
193,
215,
216,
217,
218,
219,
220,
221]. However, acute hypoxia can inhibit IRE1 and lead to reduced XBP1s levels [
222].
Finally, an elegant HIF-dependent mechanism that could result in a complete UPR activation has been proposed in endothelial cells where HIF-1 induces
VEGF through the stimulation of its receptors (VEGFRs), it activates phospholipase C (PLC), and thus, this leads to a phosphate (IP3)-dependent calcium release that initiates the UPR [
229,
230]. Furthermore, ATF6f, XBP1s, and ATF4 increase the expression of the proangiogenic genes including
VEGF, and this suggests that the UPR supports hypoxia-related angiogenesis [
231,
232,
233,
234,
235,
236,
237,
238,
239,
240,
241,
242]. In contrast, the PERK/ATF4 signals are limiting factors for erythropoietin (EPO) expression [
58].
PERK and IRE1, through their inhibitory effects on HIF-1α stability and transcriptional activity, could contribute the transition from HIF-1 to HIF-2 signaling in both endothelial and cancer cells that is observed during prolonged hypoxia, and this allows for a better adaptation to this insult [
9,
70,
88,
89]. Importantly, the HIF-mediated cellular adaptation to hypoxia relies on the induction of angiogenesis. This requires the elevated secretion of the proangiogenic factors and the increased expression of their specific transmembrane receptors as well as the remodeling of the extracellular space thorough the secreted enzymes. On one hand, all of these proteins modulate the TME, whereas on the other, these proteins need to mature properly, which requires that the ER homeostasis must be preserved [
35,
76,
275,
276,
277,
278].
5. UPR Activation in TME Cells Depends on Hypoxia Dynamics and Severity
The tumors are extremely heterogenous in terms of their microregions of oxygenation as well as the severity of the hypoxia that ranges from moderate oxygen deprivation to anoxia [72,296,297]. Furthermore, oxygen availably in the TME is often highly dynamic, and it is characterized by the periodic cycling of cells between various levels of oxygenation.
5.1. Anoxia and Extreme Acute Hypoxia
The tumor vasculature is often immature and lacks smooth muscle cells that along with it having high interstitial pressures, can result in drastic perfusion changes, including the temporary shutdown of vessels. This results in acute hypoxia or even anoxia of some small tumor regions (oxygen 0–0.1%) [
298,
299]. Notably, the tumor cells are able to survive in anoxic conditions for prolonged periods of time [
74]. Such conditions result in the complete activation of UPR signaling [
65,
146]. This includes an increase in BiP expression that is accompanied by the rapid PERK activation and translational inhibition [
188,
300], and IRE1-mediated XBP1 splicing [
146,
215,
217,
218,
219,
220,
221]. However, during the chronic exposure to acute hypoxia (above 4h), the eIF2α phosphorylation levels are a partially restored [
188,
300].
5.2. Moderate and Mild Hypoxia
The outpacing of a new blood supply compared to the rate of the tumor’s growth and the abnormal architecture of the newly formed blood vessels often have less dramatic consequences on the oxygen delivery to the tumor regions [
301,
302]. Consequently, for many of the TME cells, their oxygen availability is higher, and these cells are exposed to moderate (0.1–1% oxygen) or mild hypoxia (form 1–3%). Under these conditions, the activation of the UPR requires a longer time for it to occur, and it occurs mainly during periods of chronic exposure to hypoxia. For example, the phosphorylation of eIF2α requires more than 8 h if the oxygen concentration is moderate and consequently, a translational blockage is accompanied by the mTOR inhibition [
70].
5.3. Intermittent Hypoxia
Importantly, the transient changes in the blood flow, which are independent of the overall tumor oxygenation status result in large fluctuations in the tumor pO
2 levels that temporally increase the hypoxia severity. Such fluctuations usually can occur due to transient changes in the blood flow, and they are independent of the treatment or the overall tumor oxygenation status. As the blood flow changes from high to low, the proportion and severity of the hypoxia increases. Hence, the cells are exposed to continuous cycles of severe hypoxia, which is followed by reoxygenation. Additionally, these fluctuations can last from 30 min to 2 h [
304,
305]. Despite these relatively short period of hypoxia, this time is sufficient to switch the metabolism to the glycolytic pathway [
200,
306]. However, the restored oxygen availability will lead to the accumulation of ROS due to inability of the mitochondria to rapidly utilize “an extra” oxygen. This will then be accompanied by the rapid HIF-α degradation due to the reactivation of the PHDs, and the impairment of the HIF-related protection from the oxidative stress [
307,
308].
This entry is adapted from the peer-reviewed paper 10.3390/cancers14194870