1. Inflammation
Inflammation is a physiological defense mechanism of the immune system triggered by pathogens and damaged tissues and resolves quickly under normal circumstances. However, in autoimmune diseases, such as MS, an inflammatory misbalance leads to excessive inflammatory responses characterized by an upregulation of pro-inflammatory cytokines amplifying the inflammatory response and defective resolutive mechanisms. Autoreactive T-cells, after activation against myelin antigens, infiltrate the central nervous system (CNS), leading to inflammatory responses, which are destructive for the myelin sheath. In MS, this process is spontaneous, and its exact aetiology remains unknown [
8].
However, in the EAE models, this process is externally induced by immunization, which leads to massive inflammation against CNS. Eventually, the impaired resolution of inflammation leads to persistent chronic inflammation in the CNS. The exact pathogenesis of inflammation in the MS remains ambiguous, but its importance in the pathology is highlighted by the effectiveness of immunomodulators and immunosuppressors currently used to treat MS relapses [
9]. (
Figure 1)
1.1. Interleukin-17 (IL-17)
MS was, for a long time, considered as a Th1-mediated disease. Th0 cells are dedifferentiated in highly inflammatory Th1 cells, mainly in response to IL-12, and are characterized by the expression of pro-inflammatory cytokines such as interferon gamma (IFN-γ) [
30]. However, IL-12 KO are not only susceptible to EAE, but they also show more severe disease [
31]. Moreover, IFN-γ treatment ameliorates EAE symptoms [
32,
33]. These observations led to the identification of IL-17-producing Th17 cells as another T cell subset with a high pathogenic potential, inducing inflammation and autoimmunity. High numbers of Th17 cells are detected in the CNS of acute EAE. Similarly, high numbers of Th17 cells are present in the CSF of MS patients, especially during relapses [
34]. They attach to brain endothelial cells better than Th1 cells expressing molecules such as CCR6, enhancing their entry into the CNS [
34,
35]. They exert their effect by secreting the cytokine IL-17, although they can also sometimes express IFN-γ, depending on the tissue environment [
36]. IL-17 can be expressed not only by Th17 cells but also by astrocytes and oligodendrocytes in the CNS lesions [
37]. IL-17 plays a key role in MS inflammation. IL-17 activates and supports microglial proliferation and further promotes neuroinflammation [
38]. Indeed, IL17 expression is increased in lymphocytes derived from EAE mice, and the IL17 receptor is significantly upregulated in the CNS of EAE mice [
39]. Immunization with IL-17 conferred complete resistance to EAE [
40]. IL-17 mRNA and protein levels are increased in both brain lesions and the CSF-derived MNCs of MS patients [
41,
42]. Data from MS patients have shown that IL-17 is increased in the CSF of RRMS patients and correlates with the CSF serum albumin quotient representative of BBB disruption, implying a correlation of IL-17 in BBB disruption and indicating that targeting IL-17 could preserve BBB integrity [
43]. Indeed, IL-17 KO mice [
44] or mice administered with an anti-IL-17 antibody have shown milder EAE scores with reduced immune cell CNS infiltration, reduced inflammation and increased preservation of BBB integrity [
45]. In an in vitro study by Rahman et al., it has been demonstrated that IL-17 mediates the reorganization of actin and modifies the localization of claudin and occludin, increasing BBB permeability. In vitro studies in both human and murine brain-derived primary microvascular endothelial cells have shown that IL-17 can disrupt their integrity [
46]. As observed in IL-12 KO mice studies, anti-IL-12 or anti-IL-23 antibodies (inducers of IL-17) have failed to show any therapeutic effects in MS patients [
47]. However, the encouraging preclinical data concerning the direct blocking of IL-17 led to the use of an anti-IL17 antibody. The direct blocking of IL-17 by secukinumab in a phase II clinical trial in MS patients has shown good tolerability but a low ability to reduce MRI lesion activity [
48].
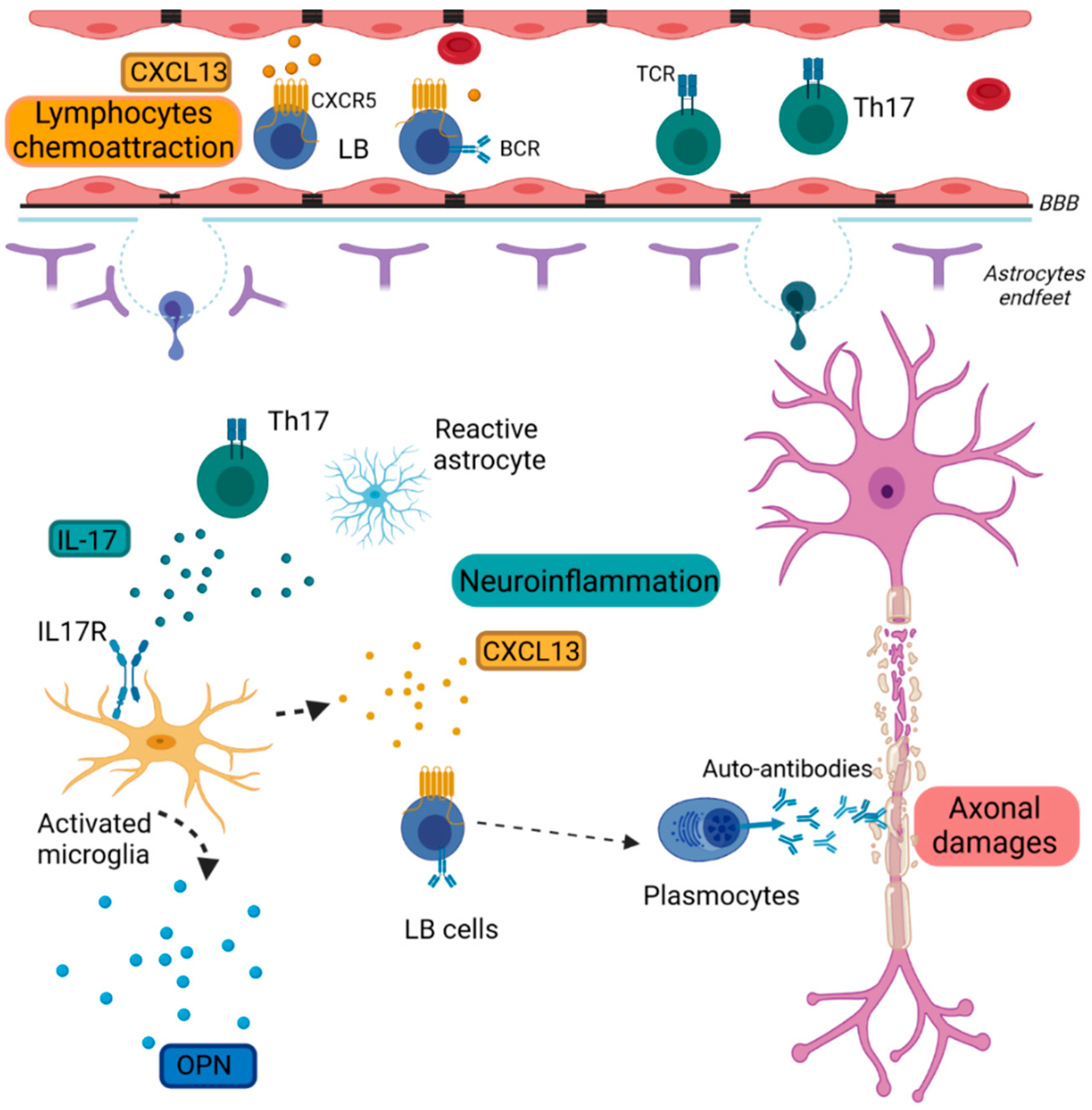
Figure 1. Schematic representation of inflammation in MS pathology.
Reactive microglia express inflammatory cytokines such as CXCL13 and OPN. CXCL13 attracts CXCR5+ B lymphocytes in the CNS. Th17 cells and reactive astrocytes secrete IL-17, which activates microglia through its receptor IL17R. B cells differentiate into plasmocytes and produce auto-antibodies targeting myelin.
BBB: Brain Blood Barrier, BCR: B-cell receptor; CXCL13: Chemokine (C-X-C Motif) Ligand 13, CXCR5: Chemokine (C-X-C Motif) receptor type 5, IL-17: Interleukin 17, IL-17R: Interleukin 17 receptor, LB: lymphocytes B, TCR: T-cell receptor, Th17: T helper 17 cells, OPN: Osteopontin
2. Blood–Brain Barrier Breakdown
The BBB is a highly regulated vasculature network consisting of endothelial cells joined by tight junctions and surrounded by astrocytic end-feet and pericytes separating the immune-privileged CNS from the systemic blood circulation. It maintains CNS homeostasis by selectively regulating the influx and efflux of solutes and immune cells in the CNS. The maintenance of the BBB functionality and dynamic interactions is crucial for proper CNS function. Changes in this regulated CNS microenvironment leading to BBB breakdown have been observed in numerous CNS disorders such as MS [
49]. The loss of BBB integrity is observed in MS lesions and is considered as the initial step in MS pathogenesis—the development of inflammatory lesions around venules which can be visualized by susceptibility-based MRI sequences [
50,
51]. It is characterized by vascular leakage, the decreased expression of tight junction proteins and the infiltration of CNS-specific immune cells. Once these cells enter the CNS, they proliferate and trigger numerous neuroinflammatory responses that result in the damage of myelin, oligodendrocytes and, ultimately, neurons. BBB has been proposed as a therapeutic target for MS to refrain leakage and immune cells infiltration in the CNS. Natalizumab, a monoclonal antibody approved for the treatment of MS, blocks the VLA-4-dependent migration of immune cells across the BBB by blocking their adhesion to VCAM-1 present on its activated endothelium. Therefore, BBB is an interesting target for the development of novel treatments promoting the restoration of the barrier function to limit immune cell attacks in the CNS, avoiding the side effects of immunosuppressive treatments [
49]. (
Figure 2)
2.1. Metalloproteinase-2 and -9 (MMP2 & MMP9)
MMPs are endopeptidases responsible for the degradation of ECM proteins. They are counteracted by tissue inhibitors of metalloproteinases (TIMPs) and play a crucial role in tissue remodeling [
52]. They are secreted, among other means, by T cells, monocytes, glial and endothelial cells [
53]. Since 1997, Chandler et al. have shown that the upregulation of MMP9 leads to tissue destruction and cellular trafficking across BBB [
52]. MMP9 CSF and serum levels correlate with the MS clinical score [
54,
55], and its expression correlates with Gd enhancements, reflective of BBB disruption [
56]. EAE mice show increased spinal cord levels of MMP-9, correlating with maximum disease severity [
57]. The selective inhibition of MMP9 resulted in an amelioration of the clinical score in an EAE model [
58]. MMP9 KO mice showed reduced BBB permeability. Mice deficient for MMP2 and MMP9 showed no BBB permeability and no brain infiltration, implying a role of both MMPs in BBB lesions [
59]. According to Agrawal et al., blood vessels are surrounded by two basement membrane barriers. The first lies outside endothelial cells, and the second lies further within CNS, adjacent to astrocytes. The inhibition of both MMPs caused the trapping of leukocytes in the space between the first and the second membrane, demonstrating the importance of MMPs for BBB disruption and auto-reactive lymphocytes’ infiltration [
60]. Nevertheless, there are conflicting results concerning the potential use of MMPs as disease biomarkers in MS patients. The MMP9/TIMP-1 ratio has been proposed as reflective of disease activity by some studies [
61,
62], but others found no correlation between MMP expression and disease activity [
63]. These differences could be explained by the fact that MMPs are regulated in several levels: gene transcription, synthesis, secretion as a proenzyme, activation, potential inhibition by TIMPs and glycosylation, which protects MMPs from degradation [
53]. Fainardi et al. studied CSF and serum MMP9 levels, detecting only active forms of MMP9. This showed that MMP9 was increased in MS compared to other inflammatory diseases, but the intrathecal synthesis represented only 18% of patients, suggesting that treatments targeting MMP9 could be beneficial only for a subgroup of patients [
64]. Nowadays, IFN-β is the first disease-modifying treatment in MS, and even though its mechanism of action is not completely elucidated, it is known that IFN-β treatment can suppress the expression of MMPs [
65]. The presence of neutralizing antibodies towards IFN-β is a cause of treatment failure. Moreover, MMP9 can indeed degrade IFN-β. Studies by Nelissen et al. showed that the proteolytic activity of MMP9 significantly reduced the bioactivity of administered and endogenous IFN-β [
66]. The treatment of patients with IFN-β—combined with tetracycline or doxycycline, antibiotics counteracting MMP9—resulted in decreased lesion activity [
67]. Overall, these results indicate a crucial role of MMP2 and MMP9 in BBB disruption and a possible correlation with IFN-β treatment failure in MS patients.
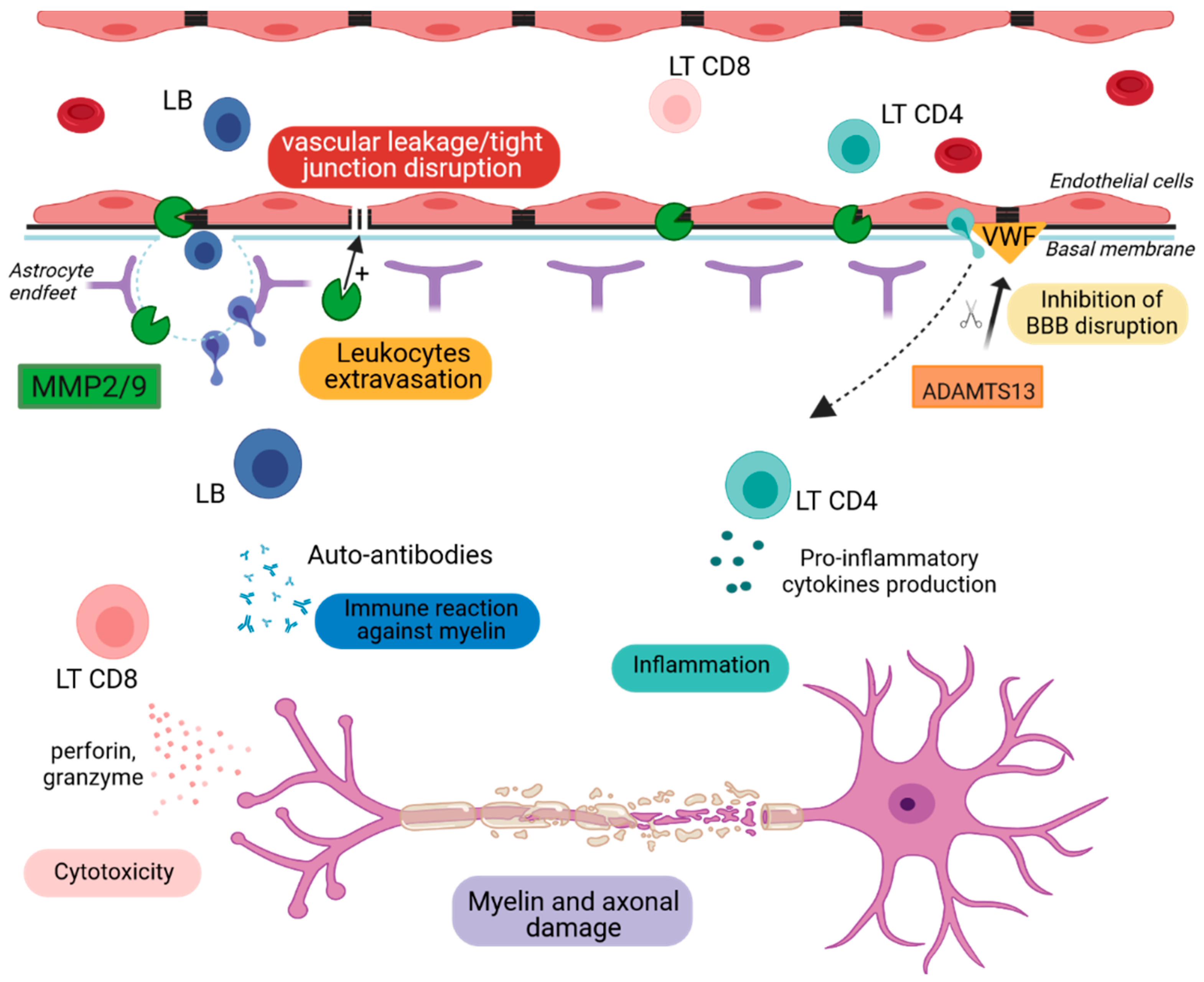
Figure 2. Schematic representation of BBB breakdown.
MMP2 and MMP9 degrade extracellular matrix proteins, leading to BBB disruption and the entry of immune cells into the CNS. LB secrete auto-antibodies that target myelin. LT CD8+ and LT CD4+ cells produce cytotoxic agents and inflammatory cytokines, respectively, that contribute to myelin destruction and cell apoptosis. ADAMTS13 cleaves the von Willebrand factor, leading to the decrease in platelet adhesion and aggregation responsible for BBB permeability.
ADAMTS13: A Disintegrin and Metalloproteinase with a Thrombospondin Type 1 Motif, Member 13, MMP2/9: Metalloproteinase-2 and -9, LB: lymphocytes B, LT CD4+: CD4 T lymphocytes, LT CD8+: CD8 T lymphocytes, VWF: Von Willebrand factor.
3. Astrogliosis
Glial cells are key cells of the CNS [
73]. There are three types of glial cells: astrocyte, oligodendrocyte and microglia. Astrocytes are the most abundant cell type in the nervous system, making up half of the brain cells, and they are mostly involved in gliotransmission, synapse development, neuronal metabolic support and long-term plasticity [
74,
75]. Increasing evidence shows the involvement of astrocytes in neurodegenerative disorders due to their aberrant dysfunction [
76]. Such deleterious mechanisms are known as reactive gliosis, mainly triggered by hypertrophic and hyperplasic astrocytes after CNS injury [
77]. This leads to the formation of glial scars corresponding to the proliferation and accumulation of fibrous astrocytes at the lesion sites. Therefore, dysfunctional astrocytes secrete inflammatory cytokines in order to attract immune cells in the lesion sites [
78]. This could constitute the first immune-related mechanism in which the astrocytes cause demyelination and contribute to the neurodegeneration. Failing in glutamate recapture induces excitotoxicity, which leads to oligodendrocyte, axonal and neuronal injuries [
79] (
Figure 3). All of the above-mentioned mechanisms inhibit the repair system.
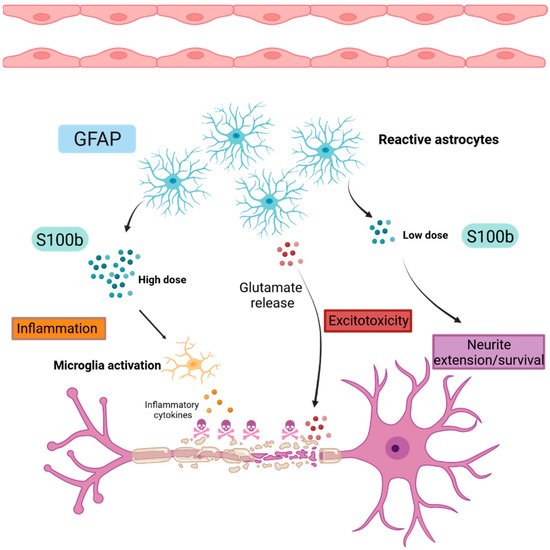
Figure 3. Schematic representation of astrogliosis.
Reactive astrocytes highly express GFAP during gliosis and produce S100B proteins that have a dual function. At low doses, S100B promotes neurite extension and neuronal survival. Inversely, at high doses, S100B activates microglia, leading to the production of inflammatory cytokines and contributing to the establishment of a neuroinflammatory environment. Reactive astrocytes release glutamate, leading to neuronal death by excitotoxicity.
GFAP: Glial Fibrillary Acidic Protein, S100b: S100 calcium binding protein B.
3.1. Glial Fibrillary Acidic Protein (GFAP)
GFAP is known to be an intermediate filament of the mature astrocytes cytoskeleton [
80]. GFAP is widely used as a CNS biomarker in several neurological disorders such as traumatic brain injury or in MS, where it is considered as a potential biomarker candidate of astrogliosis related to disease progression and severity [
81]. In EAE animal models, GFAP is upregulated at the demyelination sites during the peak of the disease [
82]. However, astrocyte contribution in pathogenesis is more arduous given their dual function. Indeed, the in vivo ablation of astrocytes worsened the disease at the early stage by allowing the proliferation of infiltrated immune cells into the CNS, causing tissue damages [
83]. Inversely, the selective ablation of reactive astrocytes at the chronic phase of the disease decreases the demyelination [
84]. This may suggest a potential protective role of the astrocytes during the early stage of the disease and a detrimental role during the late disease phase.
As the EAE model suggested, postmortem studies on patients correlate with GFAP upregulation in the lesion sites. Moreover, studies conducted in the blood and CSF of RRMS patients confirmed higher levels of GFAP compared to healthy controls, associated with larger lesions detected on MRI scans and increased disability [
85]. In addition, higher GFAP levels were found in PPMS patients compared to RRMS patients, with a stronger correlation with EDSS [
86,
87].Considering the pathological isomorphism with human disease, the research of astrogliosis biomarkers in animal models is fundamental.
3.2. S100 Calcium Binding Protein B (S100B)
S100B is a small calcium binding protein synthesized mostly by astrocytes and, to a lesser extent, by oligodendrocytes and some neuronal subpopulations. Secreted S100B can exert both neurotrophic and neurotoxic effects in a dose-dependent manner. At low doses (nM), it can promote neurite extension and survival (
Figure 3). Inversely, in stress conditions, astrocytes further secrete S100B, and its high concentration (µM) is recognized as a DAMP signal that leads to microglia activation, migration and the release of oxidative stress and proinflammatory factors that finally result in neuronal death [
88,
89]. In high concentrations, S100B can also activate nearby granulocytes and monocytes, promoting inflammation. In ex vivo demyelinating models, it has been shown that S100B levels are increased upon demyelination, with a parallel activation of astrocytes and microglia and an increased expression of inflammatory cytokines. However, the inhibition of this protein results in decreased demyelination and gliosis and the decreased expression of inflammatory molecules [
89]. Moreover, increased S100B levels impair developmental oligodendrogenesis and proper myelination in primary cultures of rat oligodendrocytes and organotypic slices, respectively. These effects are taking place after the interaction of S100B with its RAGE receptor [
90]. The elevated expression of S100B has also been observed in both active and chronic MS lesions [
85], and its receptor RAGE is strongly expressed by macrophages and microglia in active lesions [
89]. The blockage of RAGE leads to decreased demyelination in the EAE model [
91], which implies that the S100B/RAGE axis is a putative target to enhance myelination in MS. Elevated S100B levels have also been detected in the sera [
89] and the CSF samples of MS patients during relapses and the acute disease phase, while decreasing levels are observed during the remission phase of the disease and in natalizumab-treated patients [
85,
92]. In a recent study, pentamidine, an approved antiprotozoal drug and an S100B inhibitor, resulted in the amelioration of the EAE course, a reduction in immune cell infiltration and proinflammatory cytokines expression [
93].
4. Myelin and Axonal Damage
Immune cell infiltration in the CNS contributes to the onset of myelin damage [
94]. Indeed, once infiltrated in the CNS, CD4
+ T cells proliferate and produce proinflammatory cytokines that lead to the destruction of myelin sheaths [
95]. Increasing evidence suggests a role of cytotoxic CD8
+ T cells in the recognition of oligodendrocytes and myelin components. In addition, CD8
+ T cells secrete myelinotoxic agents such as perforin and granzymes, which have an immediate effect by damaging oligodendrocytes and neuronal membrane cells [
96]. Furthermore, glutamate release by reactive astrocytes leads to neuronal excitotoxicity [
97]. This inflammatory and cytotoxic microenvironment leads to the destruction of myelin sheaths and axonal damages. (
Figure 4)
4.1. Myelin and Oligodendrocyte Glycoprotein (MOG)
Myelin and oligodendrocyte glycoprotein is a minor protein component exclusively expressed in the CNS [
98,
99]. According to ultrastructure studies, MOG is expressed on the oligodendrocytes membrane and at the myelin sheath surface [
100]. Its localization made it the most common target during immune system overreaction, as in MS [
101]. MOG was first described as an autoantigen inducing intense demyelination lesions and increasing EAE severity in guinea pig, rat and mouse EAE models [
102,
103]. In addition to the encephalitogenic T cell response, antibodies directed against MOG are responsible for demyelination by targeting the component of the myelin membrane. Moreover, correlation with MOG antibodies (MOG-Ab) titers and demyelination activity was also established [
104,
105]. As a consequence, MOG became one of the most used antigens to model PPMS [
106]. The detection of MOG-Ab by ELISA in MS patients at the beginning of the 2000s was a period of huge hope in terms of biomarkers of the disease [
107]. However, MOG as a biomarker of MS fell out of favor after studies showed that MOG-Ab had a low specificity in MS patients using ELISA or western blot and was not present using cell-based assay methods. This is in addition to the emergence of a new phenotype associated with MOG-Ab called MOG-associated disorder (MOGAD) in pediatric patients with acute demyelinating encephalomyelitis and patients with a form of neuromyelitis optica known as MOG-associated disorder (MOGAD) [
108,
109,
110,
111,
112,
113]. Although MOG-Ab is not a good biomarker, MOG remains implicated in MS pathogenesis. Studies describe a blood test based on the detection of circulating-free DNA markers released in the serum from dying cells [
114]. Based on this method, MOG circulating-free DNA (cfDNA) was found in the serum of demyelinating models in high levels, which is indicative of oligodendrocyte death. A high level of demethylated MOG cfDNA was also found in RRMS patients’ serum compared to healthy controls [
115]. This interesting result may offer an opportunity to use MOG as a biomarker in order to predict disease course using a non-invasive method.
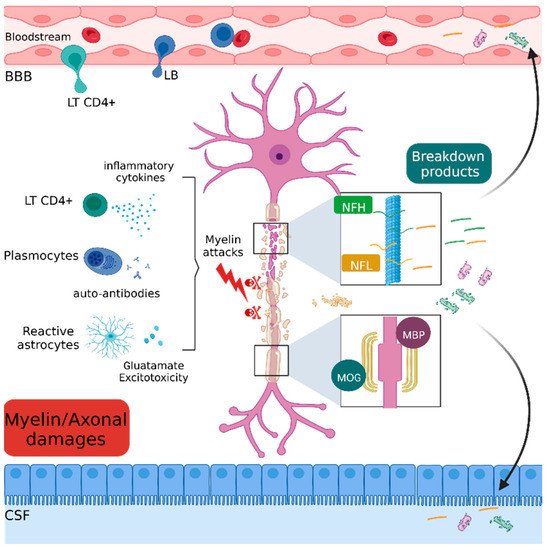
Figure 4. Schematic representation of myelin and axonal damages.
Immune, inflammatory and cytotoxic attacks lead to the destruction of axonal (NfH and NfL) and myelin (MOG and MBP) components. The residual fragments are released in the interstitial tissue and drained into the blood stream and cerebrospinal fluid.
BBB: Blood brain barrier, LT CD4+: CD4 T lymphocyte, LB: lymphocyte B, MBP: myelin binding protein, MOG: myelin and oligodendrocyte glycoprotein, NFH: neurofilament heavy chain, NFL: neurofilament light chain.
4.2. Myelin Basic Protein (MBP)
The myelin basic protein (MBP) is the second major protein in the CNS after the proteolipid protein (PLP), constituting around 30% of myelin proteins [
116]. MBP is thought to be important in the compaction of myelin sheaths during development [
117]. The lack of MBP expression in Shiverer mice leads to a thinner myelin sheath and untimely death [
118]. The same observation is made for Long Evans rats with a mutation in the MBP gene causing a loss of myelin compaction and the death of oligodendrocytes [
119]. High levels of MBP-Ig in serum are found to be discriminative between acute demyelinating encephalomyelitis and MS patients [
120]. As MS is a demyelinating disease, it is not surprising to find the MBP protein or its breakdown products in the CSF or serum. The CSF MBP levels of RRMS patients highly increase during relapses [
121], which correlates with disease activity [
122]. It is of note that MBP CSF levels are higher during relapse than progressive MS [
123]. Thus, MBP concentration could be indicative of the disease course.
4.3. Neurofilament
Neurofilament (Nf) is an intermediate filament of the neurons cytoskeleton and supports axonal architecture [
124]. Several subunits exist, depending on their different molecular weights: neurofilament light chain (NfL), neurofilament medium chain (NfM) and neurofilament heavy chain (NfH) [
125]. Nf is a hallmark of neuronal loss in several neurological diseases such as amyotrophic lateral sclerosis (ALS), Alzheimer’s, Parkinson’s disease and MS, among others [
126,
127]. In EAE mice, some studies demonstrate a strong correlation between axonal loss and a high NfH level in the serum after acute axonal injury and at the peak of the disease. Nevertheless, NfH levels decreased in chronic non-relapsing phases of the disease [
128]. This suggests that axonal injury comes directly after the first inflammatory insult, making Nf a good biomarker, reflecting an ongoing axonal injury process. In humans, the presence of elevated CSF NfL levels in a cohort of RRMS patients correlated with the EDSS, which confirms its good application as a potential biomarker [
129]. NfH levels have also been investigated, and a high CSF level was detected in a cohort of RRMS patients, as well as in CIS patients, compared to healthy controls [
130]. Moreover, experimental and meta-analysis data comparing NfL and NfH levels confirm that NfL better discriminates patients who develop MS compared to those who develop CIS [
131,
132]. Additionally, high CSF NfL levels were found in CIS patients who secondly developed an RRMS [
133]. This suggests that NfL can predict the conversion of patients with a first inflammatory episode onto patients with MS and can thereby be a prognostic biomarker. Concerning the correlation between treatment response and Nf, several studies underlined the decrease in NfL and NfH levels (serum and CSF) following treatments with natalizumab and fingolimod [
134,
135]. Further, studies showed that higher concentrations of GFAP and NfL in the serum are indicative of the severity of neurological damage [
136]. The NfL titer is now used as a secondary endpoint of neurodegeneration in phase II/III clinical trials in MS, but it is not yet in clinical use.
5. Repair
Remyelination mechanisms are characterized by the spontaneous generation of new myelin sheaths by either surviving oligodendrocytes in lesion sites or oligodendrocyte precursor cells (OPC) that migrate from distant sites. Two main steps of the remyelination are described. The first step is the recruitment of OPCs by chemoattraction mechanisms in the lesion sites due to the expression of chemoattractive molecules such as Semaphorin 3F [
137]. Following the recruitment phase and the proliferation of OPCs (through the action of platelet-derived growth factor), a second step of differentiation occurs in response to demyelination. A comprehensive review on the inhibitory factors impeding remyelination can be found in Binamé et al., 2021 [
138].
Nevertheless, the regenerative capacity of the adult CNS following myelin sheath and oligodendrocyte attacks is very poor. The remyelination failure is in part explained by (1) the impossibility of OPCs to be recruited in the lesion sites due to the overexpression of chemorepellent molecules such as Semaphorin 3A and (2) by the inhibition of OPCs differentiation and myelination through inhibitory molecules such as LINGO-1 [
139].
A neuroprotective strategy consists in the preservation of the nervous tissue including, by extension, remyelination strategies and neuronal protection. In this review, we will discuss only the neuroprotective strategies targeting pathways used by neurotrophic factors.
Two of the principal neurotrophic factors participating in MS repair mechanisms and representing potential therapeutic targets are discussed below.
5.1. Hepatocyte Growth Factor (HGF)
HGF is a pleiotropic cytokine produced by microglia, oligodendrocytes, astrocytes and neurons, among others [
140,
141]. Its receptor, the tyrosine kinase c-Met, is expressed in several tissues, including the CNS [
142]. Initially described as a mitogen for hepatocytes, HGF has demonstrated neurotrophic effects in the CNS [
143] (
Figure 5). Results in spinal cord injury (SCI) [
144] and Amyotrophic lateral sclerosis models [
145], which showed that the supplementation or overexpression of HGF delays disease progression, led to the research of potential beneficial effects in other neurodegenerative diseases. In different mouse models, HGF administration reduced disease severity by modulating both the immune response and myelin repair. Explicitly, in an EAE model, Benkoucha et al. showed that HGF overexpression resulted in decreased CNS infiltration and an increased Treg number. Bai et al., in a non-immune-mediated spinal cord lysolecithin-induced spinal cord lesion model, showed that HGF treatment improved remyelination [
140,
141]. In the CSF of MS patients, according to Müller et al., HGF concentration is significantly lower compared to healthy subjects [
146]. Its concentration is even lower in patients with active disease, and it does not seem to correlate with albumin concentration (an indicator of BBB disruption), indicating that HGF expression by CNS cells is largely reduced in MS pathology.
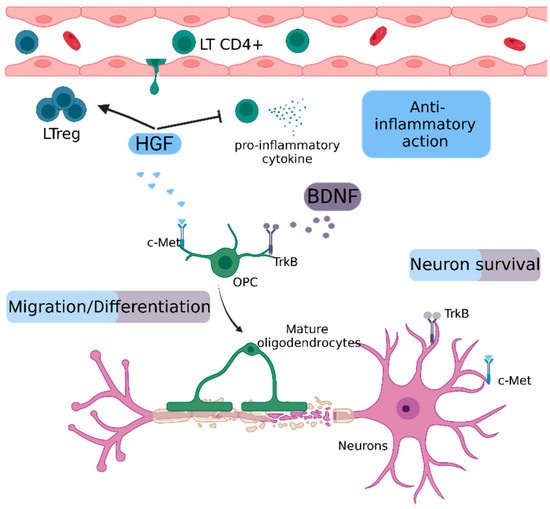
Figure 5. Schematic representation of repair mechanisms mediated by HGF and BDNF.
HGF inhibits the production of pro-inflammatory cytokines and promotes the proliferation of Treg cells. Both HGF and BDNF contribute to the migration and differentiation of oligodendrocyte precursor cells and to neuronal survival by their respective receptors, c-Met and TrkB.
BDNF: Brain-derived neurotrophic factor, c-Met: tyrosine protein kinase Met, HGF: Hepatocyte growth factor, OPC: oligodendrocyte precursor cell, LT CD4+: CD4 T Lymphocyte, LTreg: regulatory T lymphocyte, TrkB: tropomyosin receptor kinase B.
5.2. Brain-Derived Neurotrophic Factor (BDNF)
BDNF is a member of the nerve growth factor neurotrophin family, with an important role in neuron survival. BDNF plays an important role in CNS myelination, and it is involved in the recruitment, proliferation, differentiation and maturation of OPC [
147] (
Figure 5).
It is expressed by CNS components, with neurons being the main source, followed by reactive astrocytes [
148] and immune components as activated T and B cells and monocytes [
149]. Various types of neurons express BDNF in the MS brain, but immune cells are considered to be the major contributor of BDNF in MS lesions [
149]. In the EAE model, its expression in the spinal cord of EAE mice decreases during the peak of the disease [
150]. Treatment with engineered bone marrow stem cells expressing BDNF led to a milder EAE course, suggesting a potential protective effect of BDNF in EAE [
151]. In MS plasma, the results are contradictory. Some studies have shown that MS patients show decreased levels of BDNF compared to healthy patients [
152,
153,
154]. Other studies, on the contrary, have shown that the levels of BDNF increase during relapses [
155]. Although there is indisputable evidence of the neuroprotective effects of BDNF, its exogenous delivery in the CNS is difficult. A study from Kopec et al. showed that the intravenous administration of BDNF with a BBB modulator peptide enhanced BDNF delivery in the CNS and suppressed EAE relapse, promoting neuroprotective effects [
156]. This could probably pave the way for a future utilization in human disease.
This entry is adapted from the peer-reviewed paper 10.3390/ijms231911532