Intracellular calcium (Ca2+) concentration ([Ca2+]i) is a key determinant of cell fate and is implicated in carcinogenesis. Membrane ion channels are structures through which ions enter or exit the cell, depending on the driving forces. The opening of transient receptor potential vanilloid 1 (TRPV1) ligand-gated ion channels facilitates transmembrane Ca2+ and Na+ entry, which modifies the delicate balance between apoptotic and proliferative signaling pathways. Proliferation is upregulated through two mechanisms: (1) ATP binding to the G-protein-coupled receptor P2Y2, commencing a kinase signaling cascade that activates the serine-threonine kinase Akt, and (2) the transactivation of the epidermal growth factor receptor (EGFR), leading to a series of protein signals that activate the extracellular signal-regulated kinases (ERK) 1/2. The TRPV1-apoptosis pathway involves Ca2+ influx and efflux between the cytosol, mitochondria, and endoplasmic reticulum (ER), the release of apoptosis-inducing factor (AIF) and cytochrome c from the mitochondria, caspase activation, and DNA fragmentation and condensation. While proliferative mechanisms are typically upregulated in cancerous tissues, shifting the balance to favor apoptosis could support anti-cancer therapies. TRPV1, through [Ca2+]i signaling, influences cancer cell fate; therefore, the modulation of the TRPV1-enforced proliferation–apoptosis balance is a promising avenue in developing anti-cancer therapies and overcoming cancer drug resistance.
- TRPV1
- calcium signaling
- apoptosis
- proliferation
- cancers
The activation of the TRPV1 ion channel is a critical signal involved in numerous intracellular processes, some of which trigger either apoptosis or proliferation (Figure 1). While the apoptotic effects of TRPV1 are well characterized, literature on TRPV1-related proliferation remains sparse. The binding of exogenous agonists to the TRPV1 receptor and subsequent Ca2+ influx from the cytosol into the cell are characteristics shared between the apoptotic and proliferative pathways. However, both the positive allosteric modulation of cell membrane TRPV1 receptors and the activation of endoplasmic reticulum (ER)-localized TRPV1 channels are associated exclusively with the pro-apoptotic pathway [1][2].
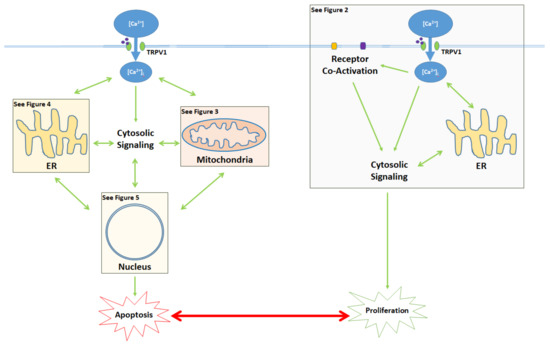
Figure 1. Activation of the TRPV1 ligand-gated ion channel causes Ca2+ influx into the cytosol and influences the balance between proliferation and apoptosis. Apoptotic signaling occurs through the cytosol, mitochondria, ER, and the nucleus. In contrast, the proliferative effects of TRPV1 are mediated by the activation of other cell membrane receptors, ER signaling, and cytosolic protein signaling cascades. The proliferative, proapoptotic mitochondrial, proapoptotic ER, and proapoptotic nuclear signaling mechanisms are highlighted in the colored boxes, and specified in Figure 2 Figure 3 Figure 4 Figure 5, respectively.
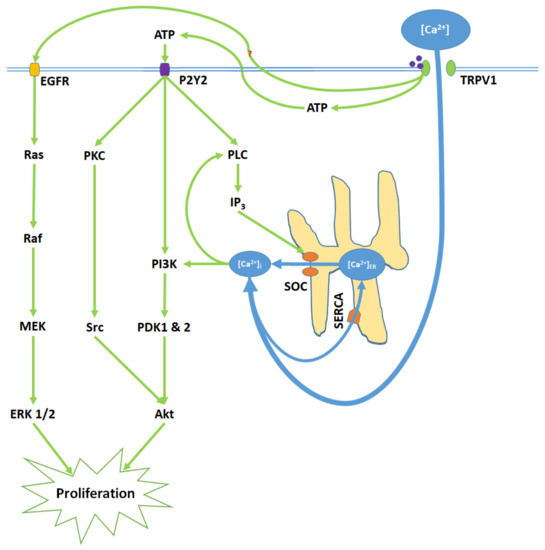
Figure 2. TRPV1 induces proliferation through Ca2+ entry, ATP release and membrane P2Y purinoceptor 2 (P2Y2) activation, and the transactivation of epidermal growth factor receptor (EGFR). Elevated [Ca2+]i and ATP-P2Y2 binding upregulate intracellular inositol triphosphate (IP3) via phospholipase C (PLC); IP3 opens store-operated channels (SOC) and thereby causes Ca2+ release from the ER. Activated P2Y2 receptors also begin the phosphoinositide-3-kinase (PI3K)/Akt pathway, a kinase signaling cascade that ultimately activates Akt. TRPV1 additionally transactivates EGFR; this prompts Ras/Raf/MAPK-ERK kinase (MEK)/extracellular signal-regulated kinase (ERK) signaling, which upregulates ERK 1/2 mitogen-activated protein kinases (MAPK). Akt and ERK 1/2 MAPK promote proliferation through nuclear activity.
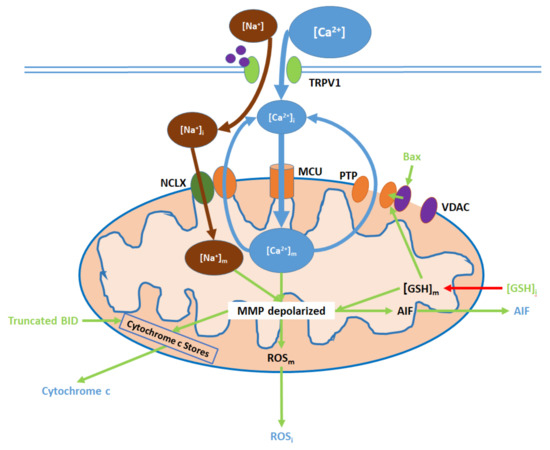
Figure 3. TRPV1 induces mitochondrial dysfunction through Ca2+ and Na+ entry, membrane depolarization, reactive oxygen species (ROS) production, and the release of cytochrome c and apoptosis-inducing factor (AIF). Initial Ca2+ and Na+ influx causes the hyperpolarization of the mitochondrial membrane, while consequent Ca2+ export through the permeability transition pore (PTP) and active Ca2+ removal via the Na+/Ca2+ exchanger (NCLX) depolarize the membrane. As inputs, downregulated intracellular glutathione ([GSH]i) and upregulated Bax arise from nuclear activity. Upon their release (driven by membrane depolarization), AIF translocates directly to the nucleus, cytochrome c participates in intracellular caspase 9 activation, and intracellular ROS (ROSi) supports the activation of p38 MAPKs.
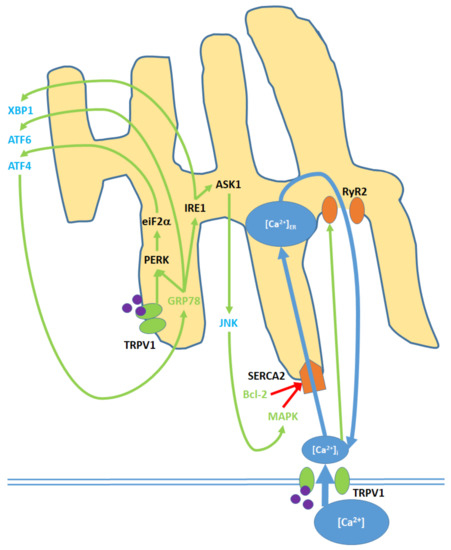
Figure 4. Cell membrane and ER TRPV1 activation promote ER stress through the modulation of [Ca2+]ER, activation of various kinases, the upregulation of nuclear transcription factors, and the release of c-Jun N-terminal kinase (JNK) into the cytosol. TRPV1 proteins localized to the ER membrane contribute only to protein signaling within the ER, while TRPV1 channels in the cell membrane promote both [Ca2+]i and protein signaling. Initial Ca2+ entry into the ER occurs through the sarco/endoplasmic reticulum Ca2+ ATPase 2 (SERCA2) pump, which is eventually blocked, causing net Ca2+ export via the ryanodine receptor 2 (RyR2) channels. Glucose regulated protein 78 (GRP78) upregulation and Bcl-2 downregulation, as inputs, arise from nuclear activity. MAPK is both an input and output of ER stress, as it is upregulated via both mitochondrial activity and JNK. Activating transcription factors 4 (ATF4) and 6 (ATF6) and X-box binding protein 1 (XBP1) are transcription factors that constitute the downstream nuclear targets of ER stress; ATF4, in particular, feeds back to the ER by upregulating GRP78.
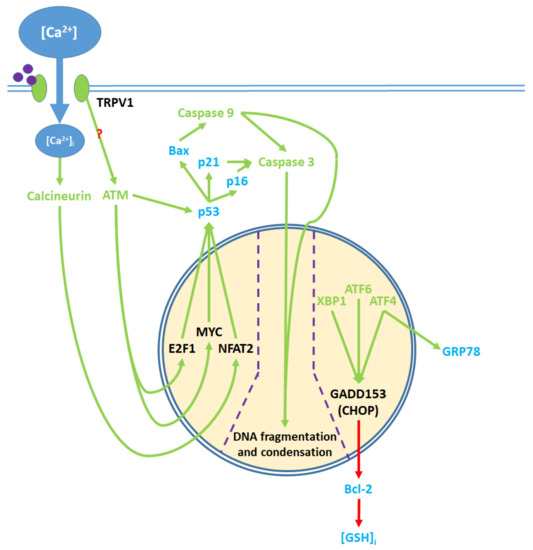
Figure 5. Pro-apoptotic processes induced by TRPV1 localized in the nucleus. The nuclear component of ER stress occurs as ATF4, ATF6, and XBP1 are activated by ER stress and upregulate GADD153, which in turn downregulates Bcl-2 protein production. The upregulation of GRP78 by ATF4 feeds back to and enhances ER stress. The cytosolic activation of the ATM serine-threonine kinase by TRPV1 protein signaling and calcineurin by elevated [Ca2+]i promote the nuclear transcription factors E2F1, MYC, and NFAT2, which upregulate p53. The precise mechanism through which TRPV1 activates ATM remains unclear. p53 upregulates the apoptotic mediators Bax, p16, and p21, which activate caspase 9 and 3. Activated caspases translocate from the cytosol to the nucleus, where they mediate DNA fragmentation and condensation.
Abbreviations: ATP, Adenosine TriPhosphate; AIF, Apoptosis Inducing Factor; Akt, Akt serine-threonine protein kinase; ASK1, Apoptosis Signal-regulating Kinase 1; ATF4, Activating Transcription Factor 4; ATF6, Activating Transcription Factor 6; ATM, ATM serine-threonine kinase; Bax, Bcl-2 associated x protein; Bcl-2, B-cell lymphoma 2; BID, BH3 Interacting-domain Death agonist; E2F1, E2F transcription factor 1; EGFR, Epidermal Growth Factor Receptor; eIF2, eukaryotic Initiation Factor 2; ERK, Extracellular signal-Regulated Kinase; GADD153, DNA Damage-Inducible Transcript 3; GRP78, Glucose Regulated Protein 78; [GSH]i, intracellular glutathione; [GSH]m, mitochondrial glutathione; IP3, Inositol triPhosphate; IRE1, Inositol-Requiring Enzyme 1; JNK, c-Jun N-terminal Kinase; MAPK, Mitogen Activated Protein Kinase; MCU, Mitochondrial Ca2+ Uniporter; MEK, MAPK-ERK Kinase; MMP, Mitochondrial Membrane Potential; MYC, Myc proto-oncogene; NCLX, Na+/Ca2+ exchanger; NFAT2, Nuclear Factor of Activated T-cells 2; P2Y2, P2Y purinoceptor 2; PDK, Phosphoinositide-Dependent Kinase; PERK, PKR-like Endoplasmic Reticulum Kinase; PI3K, PhosphoInositide-3-Kinase; PKC, Protein Kinase C; PLC, PhosphoLipase C; PTP, Permeability Transition Pore; Raf, Rapidly accelerated fibrosarcoma; Ras, Rat sarcoma; ROS, Reactive Oxygen Species; ROSi, intracellular Reactive Oxygen Species; ROSm, mitochondrial Reactive Oxygen Species; RyR2, Ryanodine Receptor 2; SERCA, Sarco/Endoplasmic Reticulum Ca2+ ATPase; SOC, Store-Operated Channel; Src, proto-oncogene tyrosine-protein kinase Src; TRPV1, Transient Receptor Potential Vanilloid 1; VDAC, Voltage Dependent Anion Channel; XBP1, X-box Binding Protein 1.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21114177
References
- Kristin Stock; Jitender Kumar; Michael Synowitz; Stefania Petrosino; Roberta Imperatore; Ewan St. John Smith; Peter Wend; Bettina Purfürst; Ulrike A. Nuber; Ulf Gurok; et al. Neural precursor cells induce cell death of high-grade astrocytomas through stimulation of TRPV1. Nature Medicine 2012, 18, 1232, 10.1038/nm.2827.
- Mustafa Nazıroğlu; Bilal Çiğ; Walter Blum; Csaba Vizler; Andrea Buhala; Annamária Marton; Róbert Katona; Katalin Josvay; Beat Schwaller; Zoltan Olah; et al. Targeting breast cancer cells by MRS1477, a positive allosteric modulator of TRPV1 channels. PLOS ONE 2017, 12, e0179950, 10.1371/journal.pone.0179950.