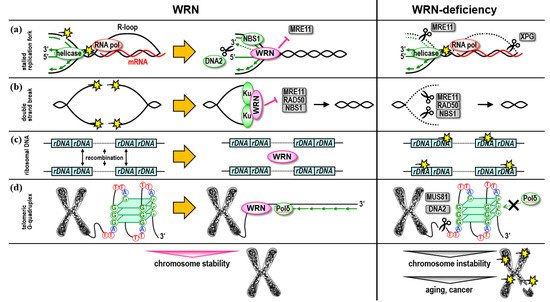
1. Role in Replication Stress Response and DNA Repair
WRN helicases localize to the nucleolus during steady-state cell cycle progression, but translocate to the site of DNA damage when it happens. WRN helicases are presumed to play a critical role in replication fork reassembly because they co-localize with RPA and Rad51 and form a focus at damaged and stalled replication forks [
11]. Recent identifications of various interaction factors have helped to illustrate their role in cellular activities. At the reversal site of the nascent DNA strand generated by the arrested replication fork, WRN is thought to support replication fork reassembly through its exonuclease activity, or that of a bound DNA2 helicase [
12,
13]. On the other hand, at the double-strand break site of the DNA generated by the arrested replication fork, WRN binds to the nascent DNA strand with NBS1, regulating the nuclease activity of MRE11 to stabilize Rad51, and supporting replication fork reconstruction by preventing the nascent DNA strand from degrading [
14]. In both cases, if the WRN does not function, small-scale DNA strand degradation occurs around the replication fork caused by other DNA repair pathways, resulting in the deletion of genetic information and abnormal chromosome structure (
Figure 1a).
Figure 1. Roles of WRN helicase in DNA repair pathways. WRN helicase is involved in the maintenance of chromosome stability through actions in DNA replication, repair, and recombination via interaction with DNA repair factors, telomere-binding proteins, and other DNA metabolic factors. (a) At the reversal site of the nascent DNA strand generated by the arrested replication fork, WRN is thought to support replication fork reassembly. At the reversal site of collision of Pol II complexes with replication forks, the cells preferentially utilize the WRN function to restore transcription and DNA replication. If the WRN does not function, small-scale DNA strand degradation occurs around the replication fork caused by other DNA repair pathways, resulting in the deletion of genetic information and abnormal chromosome structure. (b) In DNA double-strand break repair, WRN binds to the DNA ends of the breaks together with the Ku dimer, inhibits the recruitment of the MRN complex, promotes non-homologous end-joining repair, and prevents repair involving deletion of genetic information due to end resection. (c) WRN is involved in maintaining a highly regular and uniform rDNA repeat structure in the human rDNA array, suppressing illegitimate DNA recombination. (d) WRN helicase supports the synthesis of the nascent lagging strand by replicative DNA polymerase while resolving the quadruplex structure. In WS patient cells, when the replication of the nascent lagging telomere strand is stalled due to the formation of a quadruplex structure in the G-rich templates, MUS81 or DNA2 nuclease may act on the site to stimulate the resumption of replication, resulting in the shortening of the lagging telomere strand.
It is well known that WRN binds to Ku, a non-homologous end-joining (NHEJ) factor, and repairs DNA double-strand breaks other than replication forks in the NHEJ pathway [
2]. The Ku heterodimer binds to the DNA ends of double-strand breaks, and DNA repair occurs through the binding of non-homologous ends. The MRN complex (MRE11/RAD50/NBS1) is also known to bind to the DNA ends of double-strand breaks and repair them by non-homologous end joining with end resection [
2]. In the process by which either the Ku heterodimer or the MRN complex are selected to bind to the DNA ends of double-strand breaks, the WRN binds to the DNA ends of the breaks together with the Ku dimer, inhibits the recruitment of the MRN complex, promotes non-homologous end-joining repair, and prevents repair involving deletion of genetic information due to end resection [
15] (
Figure 1b).
The RNA polymerase complex, which synthesizes mRNA from chromosomal DNA, is temporarily stalled during the mRNA synthesis when it encounters DNA damage, such as DNA double-strand breaks. Marabitti et al. report that, compared with normal cells, the R-loop accumulates more in the fibroblast cells from WS patients under replication stress [
16]. The R-loop is formed when transcription complexes collide with replication forks that have been arrested under replication stress. Since the R-loop typically degrades promptly in normal cells, and transcription and replication resume [
17], WRN may play a role in the R-loop degradation. On the other hand, the degradation of the R-loop, which accumulates upon collision of Pol II complexes with replication forks in WS patient cells, is mediated by the XPG-mediated pathway responsible for the degradation of the R-loop formed by chromosomal DNA double-strand breaks [
16,
18]. These studies suggest that the cells preferentially utilize the WRN function when a specific type of DNA damage occurs. In the cells of patients with a
WRN deficiency, another pathway responds and repairs the DNA damage possibly associated with the WRN, accompanied by the deletion of genetic information and resulting abnormal chromosome structure, which in turn results in premature mitotic senescence (
Figure 1a).
2. Role in the Ribosomal DNA Regions
The ribosomal RNA gene (rDNA) locus in the eukaryotic cells consists of more than 100 copies of the same transcription unit, and such repetitive sequences are prone to recombination between the copies, which may contribute to chromosomal instability [
19]. In budding yeast, it has been shown that SIR2 suppresses undesirable recombination, and that FOB1 increases the copies to maintain an appropriate rDNA copy number and suppress cellular senescence [
19].
Sgs1 is an ortholog of the RecQ helicase with which
BLM and
WRN are functionally complementary to the gene [
20]. In a mutant strain of
Sgs1, an accumulation of arrested replication forks and DNA double-strand breaks at the ribosomal DNA replication fork barrier (RFB) was observed. Additionally, the SGS1 helicase functions in concert with DNA2 helicase and FOB1 to maintain the rDNA locus [
21].
WRN proteins have been reported to localize to the nucleolus in many cell types [
22,
23], and to migrate from the nucleolus to the nucleoplasm following certain types of DNA damage [
11,
24]. The reported role of WRN in the nucleolus is to promote transcription of ribosomal RNA by forming a complex with RNA polymerase I [
25]. However, to date, little is understood about the involvement of WRN helicase in rDNA maintenance in mammalian cells because their highly repetitive nature has hindered sequence-level analysis. We recently used Oxford Nanopore sequencing technology to show that the human rDNA array maintains a highly regular and uniform rDNA repeat structure [
26]. On the other hand, the rDNA array of WS patient cells showed an increased frequency of mutated copies, suggesting that this may be one of the causes of premature senescence [
26] (
Figure 1c). In addition, the frequency of mutations was lower in the rDNA array of the WS iPS cells compared with the original patient fibroblast cells, suggesting that cells with a stable rDNA array may have been selected during the reprogramming process [
26].
3. Role in Telomere Maintenance
The telomeres at the end of a chromosome are unique structures consisting of the 5′-(TTAGGG)n-3′ sequence repeated more than 2500 times, with specific proteins binding to the repeat sequences and protecting chromosome ends from the DNA damage response. Telomeres also play an important role in cancer suppression because they shorten during cell division in normal cells due to the terminal replication problem. It is known that the mitotic life span of the fibroblasts from WS patients is much shorter than in those of healthy individuals [
27]. Since abnormalities of telomeres have been reported in the cells derived from WS patients [
28,
29], the premature senescence observed in the cultured fibroblasts cells from WS patients may be due to abnormal telomere functions. The introduction of telomerase, a telomere-extending enzyme, into fibroblast cells from WS patients extends telomere length and prevents the premature senescence phenotype [
30]. Reprogramming of the cells by introducing Yamanaka 4 factors (i.e., Oct3/4, Sox2, Klf4, and c-myc) activates the endogenous
hTERT gene and immortalizes patient cells [
4,
5]. These results indicate that WRN helicase plays an important role in maintaining telomere structure.
Analysis using recombinant proteins has revealed that WRN helicase is an ATPase activated by single-stranded DNA with DNA unwinding activity in the 3′ to 5′ direction [
31,
32,
33], and that it performs a unique 3′ to 5′ exonuclease activity not observed in other RecQ family members [
34]. WRN helicase also performs structure-specific DNA unwinding activity and is suitable for solving G-quadruplex and recombination intermediates of Holliday structures [
35,
36,
37]. Crabbe et al. showed that WRN helicase is required for the nascent lagging strand synthesis of telomeres [
38]. Of the template strands that are unwound by the replicative helicase, the nascent lagging strand is synthesized by replicative DNA polymerase using telomere 5′-(TTAGGG)n-3′ as a template. Single-stranded G-rich telomeric DNA (i.e., TTAGGG) forms a G-quadruplex structure. It has been suggested that WRN helicase supports the synthesis of the nascent lagging strand by replicative DNA polymerase while resolving the quadruplex structure [
38]. In support of this, WRN helicase has been observed to stimulate DNA polymerase δ to proceed with DNA synthesis by overcoming G-rich repeats, and to promote the maturation of the Okazaki fragment in vitro [
39] (
Figure 1d).
On the other hand, chromosome ends are sealed by telomere-specific DNA-protein complexes, in which telomere-binding proteins, such as shelterin (TRF1, TRF2, RAP1, TIN2, TPP1, and POT1), bind to telomere sequences [
40]. Among these shelterin components, WRN helicase interacts with TRF1, TRF2, and POT1, and has been shown to play a critical role in telomere homeostasis [
41,
42,
43].
Lombard et al. report that, in vivo, mice with C-terminally truncated WRN protein without a helicase domain were viable for more than 2 years and did not show signs of premature aging [
44]. Chang et al. also examined the effect of
WRN knockout on the null background of Terc (a telomerase RNA gene) [
45]. First and second generations of
Terc-/-
Wrn-/- mice showed no apparent phenotypic alterations or shortened lifespan. On the other hand, the fourth to sixth generations of the mice with progressively shortened telomeres showed weight loss, reduced survival, premature aging typical of WS, alopecia, cataracts, and severe hypogonadism [
45]. These results show the critical importance of WRN helicase in telomere maintenance in mice with shortened telomeres.
It has been reported that the G-rich sequences that form G-quadruplexe structures are scattered in genomic regions other than telomeres at chromosome ends, and that they are involved in the regulation of gene expression [
46]. Recent studies have shown that WRN helicase plays a role in bone development and growth by targeting G-rich sequences in the promoter region of the
SHOX gene [
47], which is associated with in the proliferation and osteogenesis of chondrocytes, osteocytes, and their progenitor cells [
48], and by promoting transcription of the
SHOX gene by unwinding the G-quadruplexe structure of the promoter region [
47]. This finding clearly implicates defective
SHOX gene expression in the phenotype which causes short stature in WS patients, and suggests WRN helicase may be involved in promoting the expression of various genes by targeting G-quadruplexe structures in the genome.
4. Role in the Regulation of Heterochromatin
It is known that both the epigenomic state, represented by extensive methylation of genomic DNA, and the chromatin structure are drastically altered during cellular senescence. SAHF (senescence-associated heterochromatic foci) is a chromatin signature in senescent cells, and it is the mechanism by which proliferation-related gene expression by heterochromatin, induced by the tumor suppressor gene product, Rb protein, is suppressed [
49]. In contrast, suppressive heterochromatin in the α-satellite of the centromere and the satellite II region of the pericentromere are known to decondense and become open regions during aging [
50,
51].
Using mesenchymal stem cells (MSCs) differentiated from genome-edited human ES cells, Zhang et al. first confirmed that the passaging of cultured
WRN-deficient MSCs induced premature senescence. They found a decrease in the heterochromatin markers H3K9me3, LAP2β, and LBR in the prematurely senescent WS MSCs, which localize to heterochromatin regions around the nuclear membrane [
52]. They also identified a loss of heterochromatin regions in the pericentromere and subtelomere regions in the same cells. Because WRN helicase interacts with H3K9, trimethyltransferase SUV39H1, HP1α associated with trimethylated Lys9 of H3, and LAP2β, and because it contributes to the stabilization of heterochromatin structures in the centromere and pericentromere regions around the nuclear membrane, defective in WRN helicase leads to loss of heterochromatinization and destabilization of these regions (e.g., recombination between repeats, transcription complexes, and replication fork encounters), along with the transcriptional activation of α satellites and satellite II [
52].
There are approximately 500,000 copies of the retrotransposons called LINE-1 (L1) on the human genome, accounting for 17% of the genome. Of these, 100–150 copies are functional, encoding single-stranded endonucleases and enzymes with reverse transcriptase activity and moving across the genome [
53,
54]. In contrast, the host cells recognize retrotransposons in the genome and defend the genome by heterochromatinizing the region very early in the developmental process [
55,
56,
57]. Histone methyltransferases, such as SUV39H and SETDB1, are involved in histone H3 lysine 9 (H3K9) methylation and the formation of heterochromatin by heterochromatin protein 1 (HP1), and thus play an essential role in the heterochromatinization of retrotransposons [
58,
59].
Extensive epigenetic changes associated with senescence and aging cause retrotransposon reactivation. Decreased expression of RB1 upon cellular senescence initiates the heterochromatinization of the L1 element, and increased expression of FOXA1 promotes L1 transcription [
60]. In mice, sirtuin family members SIRT6 and SIRT7 contribute to the repression of L1 expression, and aging-induced depletion of SIRT6 and dysfunction of SIRT7 in the nuclear lamina due to loss of lamin A/C can cancel the transcriptional repression of L1 [
61,
62]. This massive L1 mRNA accumulation in the cytoplasm during aging may increase cDNA elements in the cytoplasm by reverse transcriptase activity [
63]. L1 cDNA accumulation in the cytoplasm leads to cGAS-STING-dependent type I IFN responses and an inflammatory phenotype [
60]. In SIRT6 knockout mouse fibroblasts, L1 activation and L1 cDNA accumulation in the cytoplasm cause an increased expression of type I IFN genes [
61]. These facts indicate that cytoplasmic L1 cDNA accumulation in the senescent cells maintains the inflammatory phenotype, and premature retrotransposon reactivation may trigger the inflammatory phenotype in the cells with a loss of heterochromatin due to and absence of WRN.
5. WRN Helicase as a Molecular Target for Cancer
WRN contributes to chromosome stability not only in normal cells but also in cancer cells. Thus, WRN is a feasible molecular target for anticancer drugs. We have long pursued the possibility of WRN being a potential anticancer target because we have speculated that some cancer cells depend on WRN for their survival [
64]. Recent genome-wide screening studies using CRISPR-Cas9 sgRNA libraries have reported that WRN inhibition induces synthetic lethality in cancer cells with high microsatellite instability (MSI) [
65,
66]. In the cancer cells deficient in mismatch repair, microsatellite sequences consisting of a few nucleotide repeats are unstable, and replication slippage causes large-scale elongation of the repeats to form a unique secondary structure that halts the progression of replication forks. A restart of DNA synthesis from this stalled replication fork requires unwinding of the secondary structure by the WRN, which depends on the activation of the ATR checkpoint kinase. However, when WRN is inhibited in the MSI cancer cells, the secondary structure of the elongated repeats is cleaved by the MUS81 nuclease, leading to massive chromosome disruption [
67]. After the replication fork is processed by MUS81 nuclease, the normal cells with complete DNA repair mechanisms resume replication, utilizing several repair mechanisms. On the other hand, in cancer cells, the backup of DNA repair mechanisms and cell cycle control mechanisms, including tumor suppressor genes, are defective, which leads to apoptosis with massive chromosome disruption.
In addition to MSI cancer cells, WRN helicase may be a good anti-cancer molecular target for cancer cells harboring deprotected and unstable replication forks. Indeed, a small-molecule compound that inhibits WRN helicase activity, obtained by screening, sensitizes cancer cells to some DNA-damaging agents, including topoisomerase inhibitors, poly(ADP ribose) polymerase (PARP) inhibitors, and DNA cross-linking agents [
68,
69], as well as BRCA2-mutated cells [
70]. These findings suggest that WRN helicase may function as a guardian of the genome in cancer cells in which certain DNA repair pathways are vulnerable [
71], and that WRN helicase inhibitors, such as PARP inhibitors, which act to increase genomic instability, may play an important role in the clinical development of cancer drugs [
72].
This entry is adapted from the peer-reviewed paper 10.3390/genes13101802