Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Senescence is generally considered as a process of tumor suppression, both by preventing cancer cells proliferation and inhibiting cancer progression. It can also be a key effector mechanism for many types of anticancer therapies such as chemotherapy and radiotherapy, both directly and through bioactive molecules released by senescent cells that can stimulate an immune response. There is a possibility that a combination of prosenescence therapy—which targets tumor cells and causes their senescence—with senotherapy—which targets senescent cells, can be promising in cancer treatment.
- senescence
- cancer
- therapy
- senolysis
1. Senescence and Cancer
Senescence is considered to be a process that prevents the progression and invasion of cancer cells and is exploited in anticancer therapies. Despite the ample evidence of beneficial effects of senescence in fighting cancer, there are numerous reports of its protumor activity [1][2][3]. In addition, research indicates the detrimental effects of chronic senescence not only on abnormal cells, but also on normal ones [4]. Both positive and negative effects of senescence are presented in Figure 1. Despite negatives, senescence seems to be an interesting way to fight cancer, especially when it comes to senotherapies—drugs that eliminate senescent cells or suppress their SASP. For this reason, getting acquainted with positive and negative effects that senescence and the SASP may have on cancer cells is crucial.
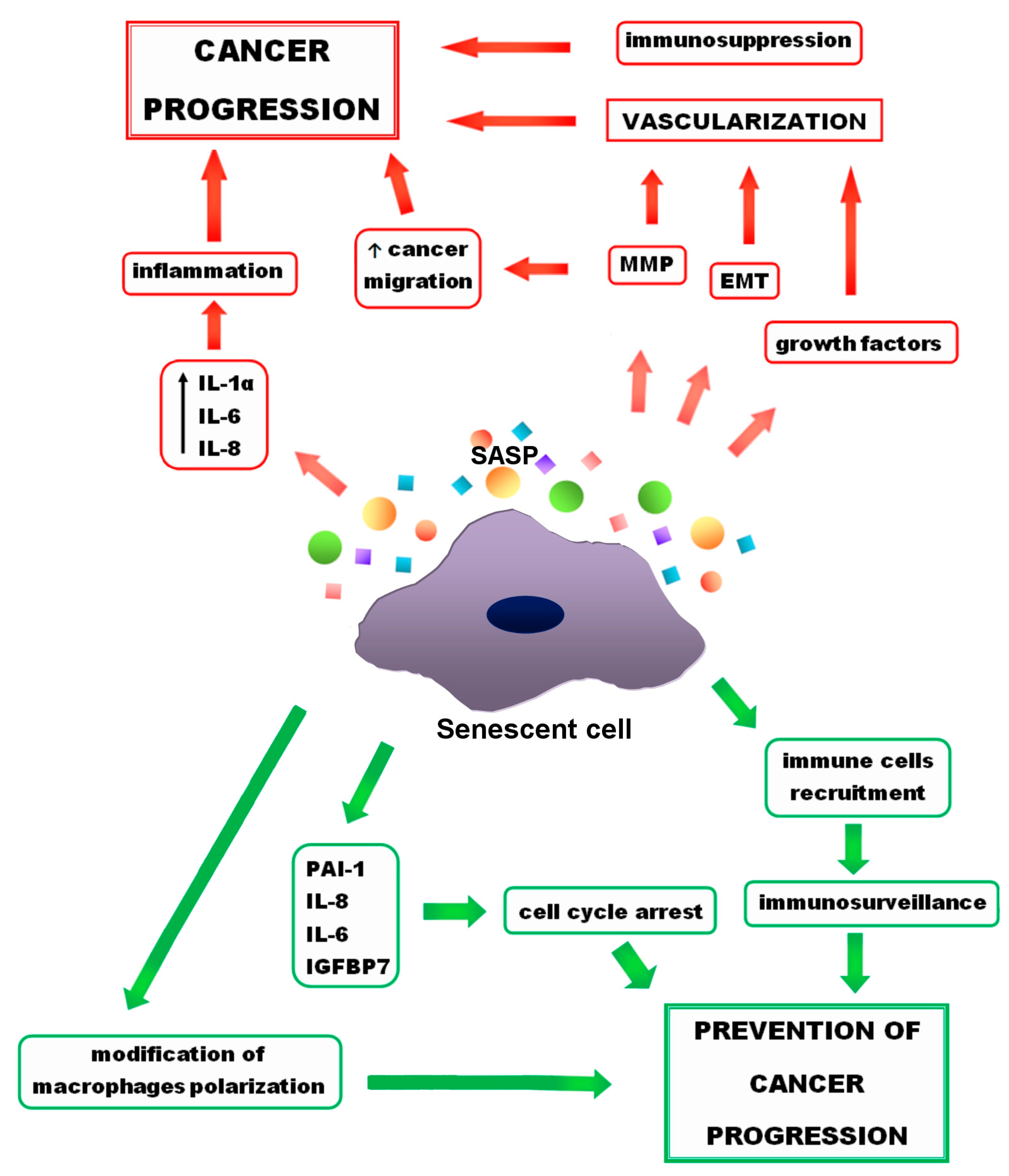
Figure 1. Positive (green arrows) and negative (red arrows) effects of senescence on cancer progression. Growth factors and metalloproteinases (MMP), released by senescent cells, as well as epithelial-to-mesenchymal transformation (EMT) lead to the cancer vascularization. MMPs also support cancer migration. Other (SASP) components, such as interleukin 1 α (IL-1 α), interleukin 6 (IL-6), interleukin 8 (IL-8) result in inflammation, which promotes cancer progression. On the other hand, senescence has also a preventive activity. Some of the SASP components, such as IL-6, IL-8, plasminogen activator inhibitor (PAI-1), insulin-like growth factor-binding protein 7 (IGFBP7) support cell cycle arrest and prevent cancer progression. Moreover, due to the senescence, immune cells recruitment and changes in macrophage polarity occur that prevent cancer progression.
1.1. Tumor Suppression
As mentioned before, senescence plays a key role in tumor suppression by enhancing immune surveillance and decreasing malignant cells proliferation. It has been proven that inactivation of senescence signaling leads to the acceleration of cancer development. A lower level of senescence in invasive cancer and higher level in premalignant lesions were observed, which support the thesis that senescence inhibits malignant progression [5][6].
The SASP consolidates cell cycle arrest via a positive-feedback loop. Inhibition of some SASP elements, such as IL-6, C-X-C motif chemokine receptor 2 (CXCR2), IGFBP7 prevents senescence, and the lack of senescence may promote cancer development [7][8][9]. IL-6 and IL8 influence SASP through CXCR2. DDR activation and enhanced ROS production occur, which enhance cell cycle arrest [7][10]. Furthermore, the SASP affects nearby normative cells in a paracrine way and induces stable cell cycle arrest, which prevents neoplastic transformation [11][12].
Immunosurveillance
Senescent cells can, through SASP, activate senescence surveillance—an anticancer immune response in order to inhibit progression of malignant cells [13]. This mechanism is associated with antigen-specific CD4(+) T cells activity and enhances an immune response to cancer tissue. Additionally, SASP contributes to NK cells recruitment and modification of macrophages polarization in order to stop tumorigenesis [14][15].
Moreover, the activity of P53 has major impact on cancer progression [16]. Restoration of its functions induces senescence and tumor suppression in sarcomas, liver carcinomas and lymphomas, and is connected with immune response (immunosurveillance) and chronic inflammation caused by SASP [17][18].
To sum up, senescence is a promising element of anticancer therapies. Stable cell cycle arrest prevents excessive proliferation and, consequently, cancer progression. Furthermore, as a result of the SASP, an enhancement in the immune system functioning occurs, which contributes to the elimination of malignant and premalignant cells.
1.2. Tumor Promotion
Besides the positive effects of senescence, there are also negative ones. The majority of anticancer treatments impact the whole organism, which leads to accumulation of senescent cells in the tumor environment [19]. The role of the SASP in creating a tumor-promoting microenvironment was described above (Section 3.3) Moreover, the accumulation may enhance a commencement and progression of some age-related chronic diseases like fibrotic, cardiovascular or neurodegenerative illnesses [20].
1.2.1. Invasiveness
Studies show that co-culturing breast cancer cells with senescent ones enhances migration through a porous membrane [21]. IL-6 and IL-8—the SASP components probably have the major impact here. It has been proven that addition of recombinant IL-8 and IL-6 enhances the preneoplastic epithelial cells invasiveness while inhibition of these interleukins decreases it [21]. MMPs also impact cancer cell invasion. Degradation of the extracellular matrix influences the permeability of capillaries, providing growth factor as well as mitogens and supporting the spread of tumors [22][23].
1.2.2. EMT
The SASP influences cancer development by promoting EMT [19]. As a result of supplying non-aggressive breast cancer cells with the medium of senescent fibroblasts, the miscellaneous features of EMT were observed, such as accelerated expression of vimentin and reduction of both E-cadherin and β-catenin [21]. What is more, incubating mesothelioma cells with senescent mesothelioma cells media (the senescence inducer was chemotherapeutic—pemetrex) leads to enhancement of EMT hallmarks, for instance vimentin upregulation [24]. This supports the thesis that EMT may be promoted in non-senescent cells by senescent tumor cells. The crucial factor of EMT genes transcription is STAT3 [19]. As already mentioned, STAT3 is influenced by the activity of IL-6 and IL-8, and studies show that these interleukins contribute to the induction of EMT in cancer cells in vitro [25][26][27].
1.2.3. Immunosenescence
Immune senescence, especially T-cells immunosenescence, is connected with immune system ageing and can contribute to tumor development by influencing immune surveillance [4]. T-cells senescence may be induced by variety of factors, such as uncontrolled inflammation or chronic T-cells stimulation [28][29]. Senescent T-cells are metabolically active and release cytokines, such as TNFα and IL-6 [28]. Furthermore, they are able to suppress the responder T-cells proliferation, which contribute to cancer progression [30]. On the other hand, senescent T-cells also play an anticancer role by influencing the fate of macrophages [31].
It has been reported that co-culturing tumor cells with premalignant senescent hepatocytes results in NK cells dysfunction and immature myeloid cells recruitment, caused by SASP, and leads to cancer progression [32]. An immunosuppressive environment reduces the ability of immune cells to invade and target neoplastic cells [33]. The research showed an age-related lung cancer progression in mice caused by myeloid-derived suppressor cells (MDSCs), the amount of which increases with age [34]. Moreover, the senescent stromal cells in the skin ageing model were sufficient to recruit MDSCs which inhibited the T-cells response and, as a result, promoted cancer development [35].
Age-related immune dysfunctions, senescent cells accumulation and weakened immunosurveillance lead to the establishment of an immunosuppressive environment, which may contribute to worse survivability of oncological patients [32][36]. The greater expression of senescence genes is connected with a shorter relapse-free period and poorer overall survival [32].
The cell cycle arrest that occurs during senescence is usually irreversible. However, under certain circumstances, and mostly in tumor cells, the re-entry into the cell cycle may take place [37][38][39][40]. It is believed that the failure of senescence may occur due to the loss of one of the crucial senescence effectors, like P53 or P16INK4A [4]. As a result, cancer progresses in an uncontrolled manner.
2. Anticancer Therapies
Cells’ entrance into senescence seems to be an inevitable part of anticancer therapies. This process may lead to cancer regression via proliferation suppression and activation of SASP-dependent immune response, as well as to cancer relapse and invasion enhancement. The link between particular cancer therapies and induction of senescence should be considered.
2.1. Chemotherapy
Chemotherapy can lead to apoptosis or senescence depending on the functionality of tumor suppressors like P16INK4A and P53 or the duration and intensity of stimuli [4]. Many chemotherapeutic agents cause DNA damage and lead to DDR, which, as mentioned before, can cause apoptosis or senescence. Higher doses of chemotherapy usually lead to cell death, whereas moderate doses lead to cell cycle arrest [4]. Due to the fact that senescent cells remain in the body and secrete SASP molecules, there are concerns that these cells may be a potential reservoir for cancer relapse. In addition, senescent cells can gain a “stem cell”-like phenotype and promote tumor development [41][42], but on the other hand, they may reinforce the immune surveillance, induce senescence of surrounding cells and consequently inhibit cancer proliferation.
It has been experimentally established that doxorubicin induces senescence in a mouse breast carcinoma model (MMTV-Wnt1). Increased P21 expression, C-C motif chemokine ligand 5 (CCL5), C-X-C motif chemokine ligand 5 (CXCL5) and eotaxin expression, as well as senescence-associated beta-galactosidase (SA-β-galactosidase) activity were observed. Those SASP factors stimulate the malignant transformation of nearby normative cells [43]. CCL5 increases the invasiveness of cancer cells by enhancing the expression of MMP9 and MMP2 and stimulates their proliferation by upregulating c-myc and cyclin D1 [44]. CXCL5 promotes metastasis by protein kinase B (AKT)/glycogen synthase kinase 3 beta (GSK3β)/β-catenin and VEGF activation [45][46]. Eotaxin contributes to tumor invasion via MMP3 activation [47]. Interestingly, elimination of doxorubicin-induced nonmalignant senescent cells limits tumor growth and relapse [41]. However, the role of senescence in chemotherapy is more complex due to the varied SASP composition, depended on cell type and senescence inducer.
2.2. Radiotherapy
Radiotherapy uses high-energy electrically charged ions to affect tumor cells through ROS and cause DNA damage, which triggers DDR [4][19]. There is a link between the dose of IR and induced DNA damage. Major damage leads to apoptosis, whereas minor damage causes senescence [4]. Information on DNA damage is detected by various proteins and transmitted to effectors and transducers of the DDR mechanism. Cell cycle arrest takes place to provide time required for DNA repair, and if this is not possible, apoptosis or senescence occur [48]. Numerous studies demonstrate that senescence in radiotherapy is dependent on P53 status [49][50]. There is evidence that securing protein participation in repair and replication of DNA influences the cell path (apoptosis or senescence) in IR-exposed tumor cells [51]. The presence of securin in colorectal cancer cells, when exposed to IR radiation, leads to an apoptosis, while its absence results in senescence [51]. The fate of glioblastoma cells after IR exposure depends on the status of PTEN. Decreased PTEN levels result in senescence and increased levels cause apoptosis [52]. IR-induced senescence in lung tumor cells is probably regulated by miR-34a [53]. These examples show that more research is needed into the factors influencing the response to radiotherapy.
Importantly, radiotherapy accelerates an immune response which increases the target cells’ immunogenicity [54]. What is more, IR-induced senescence leads to tissue fibrosis, which is a serious complication, especially in lungs [55] and is connected with skin ulceration due to radiotherapy [56]. Senotherapy may help to mitigate this response.
2.3. Prosenescence Therapies
Taking into consideration that senescence limits tumor growth, the idea was that enhancing the senescence of cancer cells could be an option in anticancer therapy. In contrast to chemotherapy, which affects both normal and cancer cells, this approach targets only tumor cells [57].
2.3.1. Telomerase Inhibition
Bearing in mind that lack of telomerase leads to telomeres shortening and consequently to replicative senescence, inhibiting telomerase appeared to be an interesting strategy for inducing senescence in cancer cells. Due to the complexity of the telomerase complex, various telomerase inhibition strategies have been developed, such as chemical telomerase inhibitors [58], inhibitors from microbial sources, antisense oligonucleotides, nucleoside and oligonucleotides [59], gene therapy constructs, molecules targeting telomerase RNA [60][61], molecules targeting telomeres and telomerase-associated proteins, and molecules targeting human telomerase reverse transcriptase (TERT) [57]. The first reported telomerase inhibitor was zidovudine (azido-2,3-dideoxythymidine or azidothymidine), which demonstrated some rate of tumor regression both alone and in combination. Better results were observed with imetelstat—an antisense oligonucleotide—which showed effectiveness in vitro against different cancers [62][63][64]. Many other telomerase-based strategies are currently in different phases of clinical trials and show interesting results [65].
2.3.2. Topoisomerase Inhibition
As mentioned previously, inhibitors of topoisomerase I and II, such as etoposide or doxorubicin, are able to induce senescence of cancer cells. The mechanism of action of these drugs contains deregulation of DNA strands religation [66][67]. It has been proven, that doxorubicin cause breaks in distal chromosomal sequences leading to telomere dysfunction [68]. Decreased levels of RB, P107, and increased levels of P130 has been observed [69]. Furthermore, there was an enhancement of the P16 and P21 proteins expression, which confirmed the arrest of the cell cycle [70][71].
2.3.3. Modulation of Cell Cycle Machinery
Cell cycle arrest is an inherent part of a senescence response. During senescence, the higher expression of CDKs inhibitors such as P15, P21WAF1/CIP1, P27, P16INK4A occurs [72][73]. Taking this into consideration, there was an idea that an enhancement of CDK inhibitors level might contribute to prosenescence therapy. CDK inhibitors prevent the phosphorylation of RB and lead to cell cycle arrest [74][75], which results in quiescence state. There is evidence that CDK4/6 inhibitors, such as ribociclib and amebaciclib, have the ability to induce the senescence [76][77]. Furthermore, the latest research on palbociclib established its ability to induce reversible senescence in ER+ breast cancer cells lines T47D and MCF7 and complete senescence in line CAMA 1. The state is assumed to be connected with mTORC1 activity, which promotes stable growth arrest in CDK4/6 inhibitor-induced senescence. Genetic depletion of a negative mTORC1 regulator—tuberous sclerosis complex 2 (TSC2)—in cell lines MCF7 and T47D, resulted in stable growth arrest after palbociclib treatment [78]. What is more, inhibition of CDK2 triggers the senescence dependent on the c-myc oncogene in different cells [79]. In conclusion, CDK inhibitors may have therapeutic importance as prosenescence factors.
2.3.4. P53 Targeting
Given the effect that P53 has on the induction of senescence, P53 activating factors seem to have significance as a prosenescence compounds. In wild-type P53 tumors, the mouse double minute—2 homolog (MDM2)/P53 interaction was inhibited, which resulted in increased P53 activity [80]. Deacetylase sirtuin 1 (SIRT1)—involved in P53 regulation via deacetylation of P53—causes degradation and ubiquitination of this protein and suppresses its activity [81][82]. It has been proven that inhibitors of SIRT1 induce senescence in tumor models [83]. In mutant P53 tumors, senescence has been triggered by small molecules restoring wild-type activity, such as MIRA-1, PRIMA-1 [84] and its analog APR-246 [85][86]. In the absence of P53, senescence has been induced by adenoviral P53 vector [87]. There is also evidence that Src family kinase and receptor tyrosine kinase (c-Kit) inhibitors such as dasatinib induce P53-mediated senescence [88].
2.3.5. Oxidative Stress
Oxidative stress and DDR are well known senescence inducers. Research indicates, that CopA3—D-type disulfide dimer peptide derived from coprisin—a defensin-like antimicrobial peptide, is able to induce cellular senescence through oxidative stress. As a result of CopA3 activity, free radicals are released and cause a DNA double-strand breaks, which lead to DDR followed by senescence. It seems possible to induce senescence with other oxidative stress inducers [89].
2.3.6. SASP Reprogramming
SASP has immense influence on the tumor environment and can affect the cancer progression in both positive and negative ways. Reprogramming SASP may be a key factor in anticancer therapies.
Inhibition of janus kinase 2 (JAK2) leads to the SASP reprogramming and reactivates the senescence immune surveillance [90]. Simvastatin, an inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMG-CoA), downregulates SASP and suppresses the SASP-mediated growth of breast cancer cells [91]. mTOR–SASP regulator inhibition suppresses SASP and its protumorigenic activity [92]. Additionally, it decreases the ability of senescent fibroblasts to enhance tumor growth. However, rapamycin (mTOR inhibitor) disrupts also the senescence surveillance and paracrine senescence—two significant factors in cancer suppression [92], which show once again the duality of SASP effects on tumor growth.
SASP reprogramming may be a promising strategy in anticancer therapies. It is essential to look for factors that mitigate the negative effects of SASP while maintaining suppressive activity. Further search for prosenescence compounds can expand therapeutic possibilities in cancer treatment, especially when it comes to senotherapies.
2.4. Elimination of Senescent Cells
2.4.1. Senotherapies
Senotherapeutics are a new group of medicaments, which restrain the senescent cells’ deleterious influence. They can be divided into categories: senolytic drugs—exterminating senescent cells, and senostatic drugs—suppressing the SASP [4]. Research on senotherapies, in particular on senolytics, indicates their therapeutic value, especially as a support to chemotherapy. It is believed, that these drugs may ameliorate treatment resilience and target tumor senescent cells. Examples of manipulating senescence in tumor cells in order to achieve therapeutic benefits are presented in Figure 2. What is more, riddance of senescent cells induced by chemotherapy alleviates many of the side effects such as cardiac functional impairments, decreased activity or fatigue in a mouse model [41]. Using quercetin and dasatinib in a mouse model in order to remove senescent cells resulted in decreased radiotherapy adverse effects, increased cardiac function and extended life expectancy [93].
Targeted elimination of senescent cells is a promising strategy in cancer treatment, especially combined with traditional methods [36]. It has been proven that quercetin has an ability to induce senolysis. The exact mechanism has not been recognised yet; however, there are reports that quercetin inhibits the phosphoinositide 3-kinase (PI3K)-AKT pathway and, as a result, disrupts the activity of the antiapoptotic protein BCL-XL [94]. Another natural flavonol with senolytic activity is fisetin, which is considered to be twice as effective as quercetin [95]. Despite the potent effectiveness of these substances in inducing senolysis, the lack of knowledge about their specific mechanism precludes their clinical use [66]. Additionally, the combination of these senolytic therapies with prosenescence therapies failed to demonstrate effectiveness in animal models of liver cancer [96].
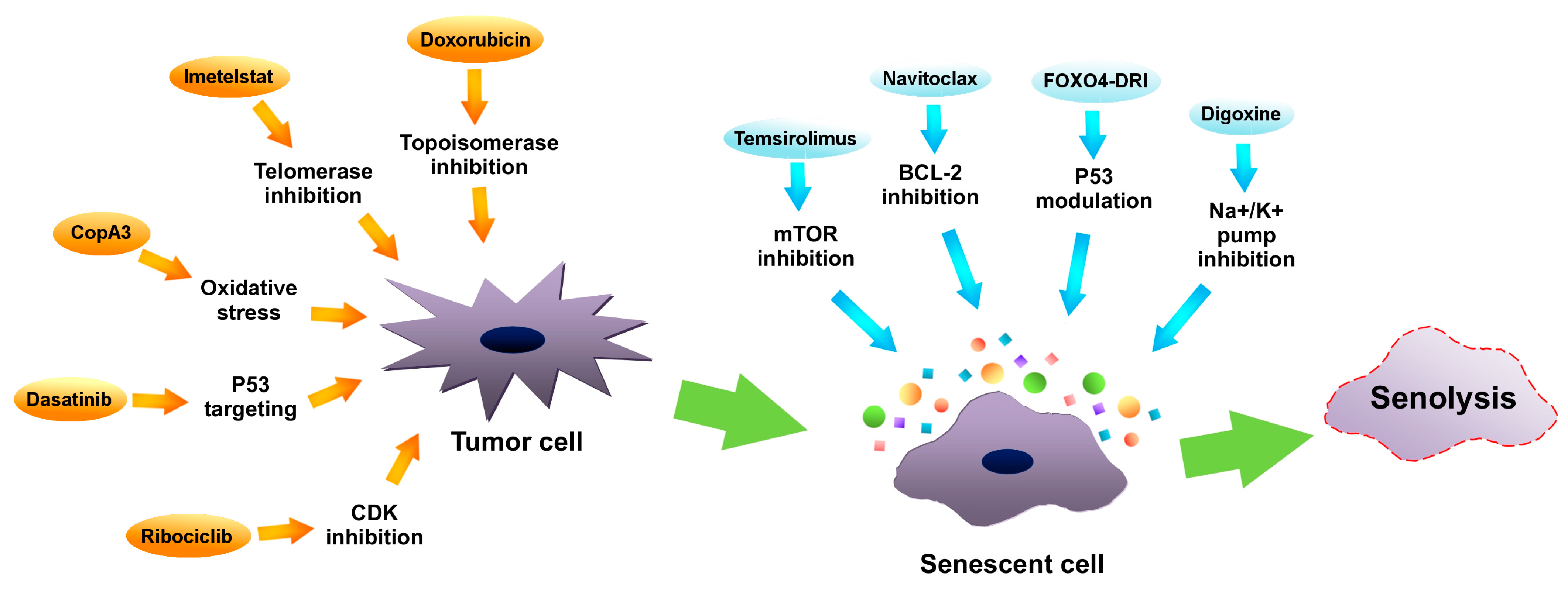
Figure 2. Manipulating senescence in order to eliminate tumor cells by senolysis. Inhibitors of telomerase (such as imetelstat), inhibitors of topoisomerase (i.e., doxorubicin), cyclin-dependent kinases (CDKs) inhibitors (like ribociclib), oxidative stress inducers (such as CopA3) as well as P53 protein modulators (i.e., dasatinib) (orange arrows) may promote tumor cells senescence. Subsequently, via using mammalian target of rapamycin (mTOR) inhibitors (such as temsirolimus), B-cell lymphoma 2 (BCL-2) inhibitors (like navitoclax), Na+/K+ pump inhibitors (such as digoxine) or P53 modulators (like FOXO4-DRI), senolysis of senescent cancer cells can be induced.
In senescent cells, a high level of BCL-2—antiapoptotic protein—has been observed [97]. The observation led to the idea of using BCL-2 inhibitors as senolytic compounds. It has been proven that navitoclax (ABT-263)—a pan-BCL-2 inhibitor, disturbs antiapoptosis associated with senescence and causes the removal of senescent cells [98]. In addition, it demonstrated an ability to exterminate senescence-like breast (MDA-MB-231) and ovarian (OV1946, OV4453) cancer cells in xenografts and culture, which were previously treated with PARP inhibitor—olaparib [99].
Research shows also that panobinostat—the histone deacetylase (HDAC) inhibitor—initiates removal of senescent cells (senolysis) of head and neck squamous carcinoma (HNSCC) and non-small-cell lung carcinoma (NSCLC) cells, pretreated with chemotherapy [100]. Moreover, research indicates the significant efficiency of cardiac glycosides, especially digoxin, in promoting senescent cancer cells senolysis, pretreated with various chemotherapeutics (doxorubicin, bleomycin, gemcitabine, palbociclib, etoposide) [101]. Cardiac glycosides inactivate the Na+/K+ ATPase pump and disturb cells’ electrochemical gradient, which leads to acidification and depolarization. Senescent cells are more vulnerable to cardiac glycosides due to their depolarized plasma membrane and increased hydrogen ion concentration [101]. Moreover, cardiac glycosides are also able to induce apoptosis through altering NOXA protein, caused by Na+/K+ transport inhibition [102].
The SASP inhibition (senostasis) may contribute to cancer therapy response improvement. Simultaneous treatment with doxorubicin and metformin (the SASP inhibitor) resulted in growth suppression of breast cancer cell xenografts in mice [103]. In addition, a similar effect was observed after rapamycin treatment in prostate cancer cell xenografts pretreated with mitoxantrone [104].
Inhibition of mTOR—a SASP regulatory factor—leads to senolysis of senescent cancer cells. The combination of the mTOR inhibitor temsirolimus (CCI-779) with chemotherapy (docetaxel) resulted in a strong antitumor activity in breast (MDA-MB-468) and prostate (PC3) xenografts and cancer cells [105]. Protein kinases are enzymes that phosphorylate other proteins in the cell and thus regulate their activity. In normal cells, the activity of protein kinases is tightly regulated, while in cancer cells it often gets out of control and is excessive. This disrupts the functioning of many cell pathways, and as a consequence leads to the intensification of cell division and uncontrolled tumor growth. Inhibition, i.e., inhibiting the excessive activity of protein kinases, is a therapeutic goal of the discussed group of anticancer drugs. Temsirolimus inhibits the activity of the mTOR protein kinase. This kinase controls not only cell division, but also the synthesis of HIF transcription factors, which regulate the tumor’s ability to adapt to oxygen deficiency conditions and produce the factor responsible for the formation of tumor vasculature (VEGF). Inhibition of mTOR kinase activity by temsirolimus limits not only the division of neoplastic cells but also lowers the level of HIF and VEGF proteins in the tumor or its environment, which inhibits the development of tumor vascularization (antiangiogenic effect) [105]. Other research showed that using XL413 in order to inhibit CDC7 (DNA-replication kinase) induced selective senescence in P53 mutated (MHCC97H, Huh7) liver cancer cells. Further addition of mTOR inhibitor (AZD8055) enhanced the tumor suppression [106]. However, disturbing the SASP does not induce senolysis every time. Cell lysis due to mTOR inhibitor is probably associated with senescent cell type [19].
Another way to induce senolysis is by modulating the activity of the P53 protein. The FOXO4-DRI peptide disrupts the P53–FOXO4 interaction, causing P53 nuclear exclusion and subsequent selective apoptosis in senescent non-cancerous cells [107]. Another P53 modulator with a senolytic activity is UBX0101, which interferes with the P53–MDM2 interaction, which results in senolysis. However, the interactions between MDM2 and P53 are not specific and can impact normal cells as well. What is more, senotherapies modulating P53 activity can only be effective in case of TP53-wild-type cancers [66].
2.4.2. Immunotherapy
Senescent cells may create a tumor-promoting microenvironment. Furthermore, they contribute to cognitive dysfunction, neurodegeneration, inflammation [108], and bone diseases such as osteoporosis or osteoarthritis [57]. Removing senescent cells from the body can alleviate the detrimental effects of senescence. Such elimination seems to be possible through immunotherapy [109][110]. Senescent cells possess on their surface different ligands, such as MHC I, MHC II, UL16 binding protein 2 (ULBP2), MHC class I chain-related protein A (MICA) and B (MICB), HLA-E, which are recognized by specific immune cells. For instance, senescent, activated stellate cells generate ligands MICA and ULBP2, which activate natural killer group 2D (NKG2D) receptor on NK cells. Different immune cells exhibit distinct ability to identify and remove specific senescent cells, which makes this strategy selective [111].
2.5. Combination of Prosenescence Therapy and Senotherapy
Despite the fact, that combining senescence promotors and senotherapeutics is still at an early stage of research, there are reports confirming the effectiveness of this strategy against cancer cells. In research conducted by Lewińska et al. [112], etoposide-induced senescent breast cancer cells (line MDA-MB-231) and senescent human normal mammary epithelial cells (HMEC) were treated with quercetin derivatives. The activity of one of them (QD3) resulted in senolysis of breast cancer cells, whereas HMEC were not affected. Research on pancreatic ductal adenocarcinoma cells [113] demonstrated effectiveness of a selective fibroblast growth factor receptor 4 (FGFR4) inhibitor—BLU9931—in the induction of senescence. Subsequently, the cells were treated with quercetin, which resulted in the death of the tumor cells. In research on prostate cancer cells [114], senescence was induced either by androgen receptor antagonist—enzalutamide (ENZ)—or by androgen receptor agonists at supraphysiological androgen level (SAL). Afterwards, cells were treated with Bcl-2 family inhibitor—ABT263, heat shock proteins (HSPs) inhibitor—ganetespib or Akt inhibitor—MK2206. While ABT263 demonstrated no senolytic activity, MK2206 induced senolysis in ENZ-treated cells. SAL-pretreated cells remained resistant to MK2206; however, they were vulnerable to ganetespib, which presented senolytic activity. In research conducted on TP53 mutant liver cancer cells [106], senescence was induced by inhibition of cell division cycle 7 (CDC7)-related protein kinase. Subsequently, cells were treated with mTOR suppressors, which caused an apoptosis of CDC7 inhibitor-treated liver cancer cells. Moreover, this combination was also applied to in vivo liver cancer models and resulted in tumor growth inhibition [106]. In research conducted by Galina et al. [115] in an immunocompetent orthotopic mouse model of the aggressive triple negative breast cancer subtype, tumor cell senescence was induced by palbociclib. The senolytic agent was nanoencapsulated navitoclax. The trial resulted in reduction of metastases, inhibition of tumor growth and reduction of systemic toxicity of navitoclax. The efficiency of combining senescence inducers and senolytics depends on variety of factors and demands further research. However, the selectivity and effectiveness of the presented approaches demonstrate the significance of these research areas for future cancer therapy.
This entry is adapted from the peer-reviewed paper 10.3390/ijms231911082
References
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077.
- Fujita, M.; Sasada, M.; Eguchi, M.; Iyoda, T.; Okuyama, S.; Osawa, T.; Tsuzuranuki, K.; Sakamoto, M.; Hagihara, Y.; Matsumura, M.; et al. Induction of cellular senescence in fibroblasts through β1-integrin activation by tenascin-C-derived peptide and its protumor effect. Am. J. Cancer Res. 2021, 11, 4364–4379.
- Wiley, C.D. Bubble Bubble, Senescent Cells Are a Cauldron of Tumor Trouble. Cancer Res. 2020, 80, 3193–3194.
- Wyld, L.; Bellantuono, I.; Tchkonia, T.; Morgan, J.; Turner, O.; Foss, F.; George, J.; Danson, S.; Kirkland, J.L. Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers 2020, 12, 2134.
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730.
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724.
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018.
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031.
- Wajapeyee, N.; Serra, R.W.; Zhu, X.; Mahalingam, M.; Green, M.R. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 2008, 132, 363–374.
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. USA 2009, 106, 17031–17036.
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990.
- Hubackova, S.; Krejcikova, K.; Bartek, J.; Hodny, Z. IL1- and TGFβ-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine ‘bystander senescence’. Aging 2012, 4, 932–951.
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551.
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-cell-autonomous tumor suppression by p53. Cell 2013, 153, 449–460.
- Iannello, A.; Thompson, T.W.; Ardolino, M.; Lowe, S.W.; Raulet, D.H. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J. Exp. Med. 2013, 210, 2057–2069.
- Yang, J.; Liu, M.; Hong, D.; Zeng, M.; Zhang, X. The Paradoxical Role of Cellular Senescence in Cancer. Front. Cell Dev. Biol. 2021, 9, 722205.
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665.
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660.
- Wang, B.; Kohli, J.; Demaria, M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer 2020, 6, 838–857.
- Campisi, J.; Andersen, J.K.; Kapahi, P.; Melov, S. Cellular senescence: A link between cancer and age-related degenerative disease? Semin. Cancer Biol. 2011, 21, 354–359.
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868.
- Liu, D.; Hornsby, P.J. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007, 67, 3117–3126.
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67.
- Canino, C.; Mori, F.; Cambria, A.; Diamantini, A.; Germoni, S.; Alessandrini, G.; Borsellino, G.; Galati, R.; Battistini, L.; Blandino, R.; et al. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene 2012, 31, 3148–3163.
- Miao, J.W.; Liu, L.J.; Huang, J. Interleukin-6-induced epithelial-mesenchymal transition through signal transducer and activator of transcription 3 in human cervical carcinoma. Int. J. Oncol. 2014, 45, 165–176.
- Goulet, C.R.; Champagne, A.; Bernard, G.; Vandal, D.; Chabaud, S.; Pouliot, F.; Bolduc, S. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of bladder cancer cells through paracrine IL-6 signalling. BMC Cancer 2019, 19, 137.
- Wang, L.; Tang, C.; Cao, H.; Li, K.; Pang, X.; Zhong, L.; Dang, W.; Tang, H.; Huang, Y.; Wei, L.; et al. Activation of IL-8 via PI3K/Akt-dependent pathway is involved in leptin-mediated epithelial-mesenchymal transition in human breast cancer cells. Cancer Biol. Ther. 2015, 16, 1220–1230.
- Vicente, R.; Mausset-Bonnefont, A.L.; Jorgensen, C.; Louis-Plence, P.; Brondello, J.M. Cellular senescence impact on immune cell fate and function. Aging Cell 2016, 15, 400–406.
- Effros, R.B.; Dagarag, M.; Valenzuela, H.F. In vitro senescence of immune cells. Exp. Gerontol. 2003, 38, 1243–1249.
- Montes, C.L.; Chapoval, A.I.; Nelson, J.; Orhue, V.; Zhang, X.; Schulze, D.H.; Strome, S.E.; Gastman, B.R. Tumor-induced senescent T cells with suppressor function: A potential form of tumor immune evasion. Cancer Res. 2008, 68, 870–879.
- Ramello, M.C.; Tosello Boari, J.; Canale, F.P.; Mena, H.A.; Negrotto, S.; Gastman, B.; Gruppi, A.; Acosta Rodríguez, E.V.; Montes, C.L. Tumor-induced senescent T cells promote the secretion of pro-inflammatory cytokines and angiogenic factors by human monocytes/macrophages through a mechanism that involves Tim-3 and CD40L. Cell Death Dis. 2014, 5, e1507.
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547.
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217.
- Chen, S.; Liu, H.; Su, N.; Zhang, G.; Wang, L. Myeloid-derived suppressor cells promote age-related increase of lung cancer growth via B7-H1. Exp. Gerontol. 2015, 61, 84–91.
- Ruhland, M.K.; Loza, A.J.; Capietto, A.H.; Luo, X.; Knolhoff, B.L.; Flanagan, K.C.; Belt, B.A.; Alspach, E.; Leahy, K.; Luo, J.; et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun. 2016, 7, 11762.
- Sieben, C.J.; Sturmlechner, I.; van de Sluis, B.; van Deursen, J.M. Two-Step Senescence-Focused Cancer Therapies. Trends Cell Biol. 2018, 28, 723–737.
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100.
- Galanos, P.; Vougas, K.; Walter, D.; Polyzos, A.; Maya-Mendoza, A.; Haagensen, E.J.; Kokkalis, A.; Roumelioti, F.M.; Gagos, S.; Tzetis, M.; et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell Biol. 2016, 18, 777–789.
- Patel, P.L.; Suram, A.; Mirani, N.; Bischof, O.; Herbig, U. Derepression of hTERT gene expression promotes escape from oncogene-induced cellular senescence. Proc. Natl. Acad. Sci. USA 2016, 113, E5024–E5033.
- Saleh, T.; Tyutyunyk-Massey, L.; Gewirtz, D.A. Tumor Cell Escape from Therapy-Induced Senescence as a Model of Disease Recurrence after Dormancy. Cancer Res. 2019, 79, 1044–1046.
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176.
- Zhang, B.; Lam, E.W.; Sun, Y. Senescent cells: A new Achilles’ heel to exploit for cancer medicine? Aging Cell 2019, 18, e12875.
- Jackson, J.G.; Pant, V.; Li, Q.; Chang, L.L.; Quintás-Cardama, A.; Garza, D.; Tavana, O.; Yang, P.; Manshouri, T.; Li, Y.; et al. p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell 2012, 21, 793–806.
- Aldinucci, D.; Colombatti, A. The inflammatory chemokine CCL5 and cancer progression. Mediat. Inflamm. 2014, 2014, 292376.
- Chen, C.; Xu, Z.Q.; Zong, Y.P.; Ou, B.C.; Shen, X.H.; Feng, H.; Zheng, M.H.; Zhao, J.K.; Lu, A.G. CXCL5 induces tumor angiogenesis via enhancing the expression of FOXD1 mediated by the AKT/NF-κB pathway in colorectal cancer. Cell Death Dis. 2019, 10, 178.
- Zhao, J.; Ou, B.; Han, D.; Wang, P.; Zong, Y.; Zhu, C.; Liu, D.; Zheng, M.; Sun, J.; Feng, H.; et al. Tumor-derived CXCL5 promotes human colorectal cancer metastasis through activation of the ERK/Elk-1/Snail and AKT/GSK3β/β-catenin pathways. Mol. Cancer 2017, 16, 70.
- Zhu, F.; Liu, P.; Li, J.; Zhang, Y. Eotaxin-1 promotes prostate cancer cell invasion via activation of the CCR3-ERK pathway and upregulation of MMP-3 expression. Oncol. Rep. 2014, 31, 2049–2054.
- Mongiardi, M.P.; Pellegrini, M.; Pallini, R.; Levi, A.; Falchetti, M.L. Cancer Response to Therapy-Induced Senescence: A Matter of Dose and Timing. Cancers 2021, 13, 484.
- Jones, K.R.; Elmore, L.W.; Jackson-Cook, C.; Demasters, G.; Povirk, L.F.; Holt, S.E.; Gewirtz, D.A. p53-Dependent accelerated senescence induced by ionizing radiation in breast tumour cells. Int. J. Radiat. Biol. 2005, 81, 445–458.
- Quick, Q.A.; Gewirtz, D.A. An accelerated senescence response to radiation in wild-type p53 glioblastoma multiforme cells. J. Neurosurg. 2006, 105, 111–118.
- Jallepalli, P.V.; Waizenegger, I.C.; Bunz, F.; Langer, S.; Speicher, M.R.; Peters, J.M.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Securin is required for chromosomal stability in human cells. Cell 2001, 105, 445–457.
- Lee, J.J.; Kim, B.C.; Park, M.J.; Lee, Y.S.; Kim, Y.N.; Lee, B.L.; Lee, J.S. PTEN status switches cell fate between premature senescence and apoptosis in glioma exposed to ionizing radiation. Cell Death Differ. 2011, 18, 666–677.
- He, X.; Yang, A.; McDonald, D.G.; Riemer, E.C.; Vanek, K.N.; Schulte, B.A.; Wang, G.Y. MiR-34a modulates ionizing radiation-induced senescence in lung cancer cells. Oncotarget 2017, 8, 69797–69807.
- Wennerberg, E.; Vanpouille-Box, C.; Bornstein, S.; Yamazaki, T.; Demaria, S.; Galluzzi, L. Immune recognition of irradiated cancer cells. Immunol. Rev. 2017, 280, 220–230.
- He, Y.; Thummuri, D.; Zheng, G.; Okunieff, P.; Citrin, D.E.; Vujaskovic, Z.; Zhou, D. Cellular senescence and radiation-induced pulmonary fibrosis. Transl. Res. 2019, 209, 14–21.
- Wang, H.; Wang, Z.; Huang, Y.; Zhou, Y.; Sheng, X.; Jiang, Q.; Wang, Y.; Luo, P.; Luo, M.; Shi, C. Senolytics (DQ) Mitigates Radiation Ulcers by Removing Senescent Cells. Front. Oncol. 2020, 9, 1576.
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078.
- Shammas, M.A.; Simmons, C.G.; Corey, D.R.; Shmookler Reis, R.J. Telomerase inhibition by peptide nucleic acids reverses ‘immortality’ of transformed human cells. Oncogene 1999, 18, 6191–6200.
- Hájek, M.; Matulová, N.; Votruba, I.; Holý, A.; Tloust’ová, E. Inhibition of human telomerase by diphosphates of acyclic nucleoside phosphonates. Biochem. Pharmacol. 2005, 70, 894–900.
- Ji, X.M.; Xie, C.H.; Fang, M.H.; Zhou, F.X.; Zhang, W.J.; Zhang, M.S.; Zhou, Y.F. Efficient inhibition of human telomerase activity by antisense oligonucleotides sensitizes cancer cells to radiotherapy. Acta Pharmacol. Sin. 2006, 27, 1185–1191.
- Kondo, Y.; Kondo, S. Telomerase RNA inhibition using antisense oligonucleotide against human telomerase RNA linked to a 2′,5′-oligoadenylate. Methods Mol. Biol. 2007, 405, 97–112.
- Burchett, K.M.; Yan, Y.; Ouellette, M.M. Telomerase inhibitor Imetelstat (GRN163L) limits the lifespan of human pancreatic cancer cells. PLoS ONE 2014, 9, e85155.
- Djojosubroto, M.W.; Chin, A.C.; Go, N.; Schaetzlein, S.; Manns, M.P.; Gryaznov, S.; Harley, C.B.; Rudolph, K.L. Telomerase antagonists GRN163 and GRN163L inhibit tumor growth and increase chemosensitivity of human hepatoma. Hepatology 2005, 42, 1127–1136.
- Marian, C.O.; Cho, S.K.; McEllin, B.M.; Maher, E.A.; Hatanpaa, K.J.; Madden, C.J.; Mickey, B.E.; Wright, W.E.; Shay, J.W.; Bachoo, R.M. The telomerase antagonist, imetelstat, efficiently targets glioblastoma tumor-initiating cells leading to decreased proliferation and tumor growth. Clin. Cancer Res. 2010, 16, 154–163.
- Relitti, N.; Saraswati, P.A.; Federico, S.; Khan, T.; Brindisi, M.; Zisterer, D.; Brogi, S.; Gemma, S.; Butini, S.; Campiani, G. Telomerase-based Cancer Therapeutics: A Review on their Clinical Trials. Curr. Top. Med. Chem. 2020, 20, 433–457.
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 2022, 22, 340–355.
- Milczarek, M. The Premature Senescence in Breast Cancer Treatment Strategy. Cancers 2020, 12, 1815.
- Elmore, L.W.; Rehder, C.W.; Di, X.; McChesney, P.A.; Jackson-Cook, C.K.; Gewirtz, D.A.; Holt, S.E. Adriamycin-induced senescence in breast tumor cells involves functional p53 and telomere dysfunction. J. Biol. Chem. 2002, 277, 35509–35515.
- Jackson, J.G.; Pereira-Smith, O.M. Primary and Compensatory Roles for RB Family Members at Cell Cycle Gene Promoters That Are Deacetylated and Downregulated in Doxorubicin-Induced Senescence of Breast Cancer Cells. Mol. Cell Biol. 2006, 26, 2501–2510.
- Inao, T.; Kotani, H.; Iida, Y.; Kartika, I.D.; Okimoto, T.; Tanino, R.; Harada, M. Different sensitivities of senescent breast cancer cells to immune cell-mediated cytotoxicity. Cancer Sci. 2019, 110, 2690–2699.
- Bojko, A.; Czarnecka-Herok, J.; Charzynska, A.; Dabrowski, M.; Sikora, E. Diversity of the Senescence Phenotype of Cancer Cells Treated with Chemotherapeutic Agents. Cells 2019, 8, 1501.
- Lin, A.W.; Barradas, M.; Stone, J.C.; van Aelst, L.; Serrano, M.; Lowe, S.W. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998, 12, 3008–3019.
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602.
- Capparelli, C.; Guido, C.; Whitaker-Menezes, D.; Bonuccelli, G.; Balliet, R.; Pestell, T.G.; Goldberg, A.F.; Pestell, R.G.; Howell, A.; Sneddon, S.; et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle 2012, 11, 2285–2302.
- Leontieva, O.V.; Blagosklonny, M.V. CDK4/6-inhibiting drug substitutes for p21 and p16 in senescence: Duration of cell cycle arrest and MTOR activity determine geroconversion. Cell Cycle 2013, 12, 3063–3069.
- Tao, Y.F.; Wang, N.N.; Xu, L.X.; Li, Z.H.; Li, X.L.; Xu, Y.Y.; Fang, F.; Li, M.; Qian, G.H.; Li, Y.H.; et al. Molecular mechanism of G1 arrest and cellular senescence induced by LEE011, a novel CDK4/CDK6 inhibitor, in leukemia cells. Cancer Cell Int. 2017, 17, 35.
- Yoshida, A.; Diehl, J.A. CDK4/6 inhibitor: From quiescence to senescence. Oncoscience 2015, 2, 896–897.
- Maskey, R.S.; Wang, F.; Lehman, E.; Wang, Y.; Emmanuel, N.; Zhong, W.; Jin, G.; Abraham, R.T.; Arndt, K.T.; Myers, J.S.; et al. Sustained mTORC1 activity during palbociclib-induced growth arrest triggers senescence in ER+ breast cancer cells. Cell Cycle 2021, 20, 65–80.
- Campaner, S.; Doni, M.; Hydbring, P.; Verrecchia, A.; Bianchi, L.; Sardella, D.; Schleker, T.; Perna, D.; Tronnersjö, S.; Murga, M.; et al. Cdk2 suppresses cellular senescence induced by the c-myc oncogene. Nat. Cell Biol. 2010, 12, 54–59.
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848.
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159.
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148.
- van Leeuwen, I.; Lain, S. Sirtuins and p53. Adv. Cancer Res. 2009, 102, 171–195.
- Qiang, W.; Jin, T.; Yang, Q.; Liu, W.; Liu, S.; Ji, M.; He, N.; Chen, C.; Shi, B.; Hou, P. PRIMA-1 selectively induces global DNA demethylation in p53 mutant-type thyroid cancer cells. J. Biomed. Nanotechnol. 2014, 10, 1249–1258.
- Bykov, V.J.N.; Issaeva, N.; Zache, N.; Shilov, A.; Hultcrantz, M.; Bergman, J.; Selivanova, G.; Wiman, K.G. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J. Biol. Chem. 2017, 292, 19607.
- Mohell, N.; Alfredsson, J.; Fransson, Å.; Uustalu, M.; Byström, S.; Gullbo, J.; Hallberg, A.; Bykov, V.J.; Björklund, U.; Wiman, K.G. APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 2015, 6, e1794.
- Shi, J.; Zheng, D. An update on gene therapy in China. Curr. Opin. Mol. Ther. 2009, 11, 547–553.
- Dos Santos, C.; McDonald, T.; Ho, Y.W.; Liu, H.; Lin, A.; Forman, S.J.; Kuo, Y.H.; Bhatia, R. The Src and c-Kit kinase inhibitor dasatinib enhances p53-mediated targeting of human acute myeloid leukemia stem cells by chemotherapeutic agents. Blood 2013, 122, 1900–1913.
- Dey, D.K.; Kang, S.C. CopA3 peptide induces permanent cell-cycle arrest in colorectal cancer cells. Mech. Ageing Dev. 2021, 196, 111497.
- Toso, A.; Revandkar, A.; Di Mitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014, 9, 75–89.
- Liu, S.; Uppal, H.; Demaria, M.; Desprez, P.Y.; Campisi, J.; Kapahi, P. Simvastatin suppresses breast cancer cell proliferation induced by senescent cells. Sci. Rep. 2015, 5, 17895.
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217.
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658.
- Paez-Ribes, M.; González-Gualda, E.; Doherty, G.J.; Muñoz-Espín, D. Targeting senescent cells in translational medicine. EMBO Mol. Med. 2019, 11, e10234.
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. eBioMedicine 2018, 36, 18–28.
- Kovacovicova, K.; Skolnaja, M.; Heinmaa, M.; Mistrik, M.; Pata, P.; Pata, I.; Bartek, J.; Vinciguerra, M. Senolytic Cocktail Dasatinib + Quercetin (D + Q) Does Not Enhance the Efficacy of Senescence-Inducing Chemotherapy in Liver Cancer. Front. Oncol. 2018, 8, 459.
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190.
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83.
- Fleury, H.; Malaquin, N.; Tu, V.; Gilbert, S.; Martinez, A.; Olivier, M.A.; Sauriol, A.; Communal, L.; Leclerc-Desaulniers, K.; Carmona, E.; et al. Exploiting interconnected synthetic lethal interactions between PARP inhibition and cancer cell reversible senescence. Nat. Commun. 2019, 10, 2556.
- Samaraweera, L.; Adomako, A.; Rodriguez-Gabin, A.; McDaid, H.M. A Novel Indication for Panobinostat as a Senolytic Drug in NSCLC and HNSCC. Sci. Rep. 2017, 7, 1900.
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M.; et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019, 10, 4731.
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 2019, 1, 1074–1088.
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. 2011, 71, 3196–3201.
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061.
- Fung, A.S.; Wu, L.; Tannock, I.F. Concurrent and sequential administration of chemotherapy and the Mammalian target of rapamycin inhibitor temsirolimus in human cancer cells and xenografts. Clin. Cancer Res. 2009, 15, 5389–5395.
- Wang, C.; Vegna, S.; Jin, H.; Benedict, B.; Lieftink, C.; Ramirez, C.; de Oliveira, R.L.; Morris, B.; Gadiot, J.; Wang, W.; et al. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature 2019, 574, 268–272.
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e16.
- Ogrodnik, M.; Evans, S.A.; Fielder, E.; Victorelli, S.; Kruger, P.; Salmonowicz, H.; Weigand, B.M.; Patel, A.D.; Pirtskhalava, T.; Inman, C.L.; et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 2021, 20, e13296.
- Sagiv, A.; Biran, A.; Yon, M.; Simon, J.; Lowe, S.W.; Krizhanovsky, V. Granule exocytosis mediates immune surveillance of senescent cells. Oncogene 2013, 32, 1971–1977.
- Burton, D.G.A.; Stolzing, A. Cellular senescence: Immunosurveillance and future immunotherapy. Ageing Res. Rev. 2018, 43, 17–25.
- Song, P.; An, J.; Zou, M.H. Immune Clearance of Senescent Cells to Combat Ageing and Chronic Diseases. Cells 2020, 9, 671.
- Lewińska, A.; Przybylski, P.; Adamczyk-Grochala, J.; Błoniarz, D.; Litwinienko, G.; Wnuk, M. Senolysis-Based Elimination of Chemotherapy-Induced Senescent Breast Cancer Cells by Quercetin Derivative with Blocked Hydroxy Groups. Cancers 2022, 14, 605.
- Sasaki, N.; Gomi, F.; Yoshimura, H.; Yamamoto, M.; Matsuda, Y.; Michishita, M.; Hatakeyama, H.; Kawano, Y.; Toyoda, M.; Korc, M.; et al. FGFR4 Inhibitor BLU9931 Attenuates Pancreatic Cancer Cell Proliferation and Invasion While Inducing Senescence: Evidence for Senolytic Therapy Potential in Pancreatic Cancer. Cancers 2020, 12, 2976.
- Pungsrinont, T.; Sutter, M.F.; Ertingshausen, M.C.C.M.; Lakshmana, G.; Kokal, M.; Khan, A.S.; Baniahmad, A. Senolytic compounds control a distinct fate of androgen receptor agonist- and antagonist-induced cellular senescent LNCaP prostate cancer cells. Cell Biosci. 2020, 10, 59.
- Galiana, I.; Lozano-Torres, B.; Sancho, M.; Alfonso, M.; Bernardos, A.; Bisbal, V.; Serrano, M.; Martínez-Máñez, R.; Orzáez, M. Preclinical antitumor efficacy of senescence-inducing chemotherapy combined with a nanoSenolytic. J. Control. Release 2020, 323, 624–634.
This entry is offline, you can click here to edit this entry!